 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4hgu: Crystal Structure of Galleria mellonella Silk Protease Inhibitor 2 -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4hgu | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of Galleria mellonella Silk Protease Inhibitor 2 | ||||||
 Components Components | Silk protease inhibitor 2 | ||||||
 Keywords Keywords | HYDROLASE INHIBITOR / KAZAL-TYPE SERINE PROTEASE INHIBITOR | ||||||
| Function / homology |  Function and homology information Function and homology informationKazal-type serine protease inhibitor domain / Wheat Germ Agglutinin (Isolectin 2); domain 1 - #30 / Kazal type serine protease inhibitors / Kazal domain superfamily / Kazal domain / Kazal domain profile. / Wheat Germ Agglutinin (Isolectin 2); domain 1 / 2-Layer Sandwich / Alpha Beta Similarity search - Domain/homology | ||||||
| Biological species |  Galleria mellonella (greater wax moth) Galleria mellonella (greater wax moth) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 0.98 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 0.98 Å | ||||||
 Authors Authors | Krzywda, S. / Jaskolski, M. / Dvornyk, A. / Kludkiewicz, B. / Grzelak, K. / Zagorski, W. / Bal, W. / Kopera, E. | ||||||
 Citation Citation | Journal: Plos One / Year: 2014 Title: Atomic resolution structure of a protein prepared by non-enzymatic His-tag removal. Crystallographic and NMR study of GmSPI-2 inhibitor. Authors: Kopera, E. / Bal, W. / Lenarcic Zivkovic, M. / Dvornyk, A. / Kludkiewicz, B. / Grzelak, K. / Zhukov, I. / Zagorski-Ostoja, W. / Jaskolski, M. / Krzywda, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4hgu.cif.gz | 34.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4hgu.ent.gz | 22.3 KB | Display | PDB format |
| PDBx/mmJSON format | 4hgu.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hg/4hguftp://data.pdbj.org/pub/pdb/validation_reports/hg/4hgu | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2m5xC  1an1S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 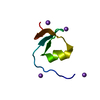
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein/peptide | Mass: 4317.744 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Galleria mellonella (greater wax moth) / Plasmid: pPICZalphaB / Production host:  Pichia pastoris (fungus) / References: UniProt: Q968S7 Pichia pastoris (fungus) / References: UniProt: Q968S7 | ||||
|---|---|---|---|---|---|
| #2: Chemical | ChemComp-NA /   Mass: 22.990 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Na#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 81 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 81 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.76 Å3/Da / Density % sol: 30.23 % |
|---|---|
| Crystal grow | Temperature: 292 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: 1.4 M sodium citrate and 0.1 M Hepes pH 7.5, VAPOR DIFFUSION, HANGING DROP, temperature 292K |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: BESSY  / Beamline: 14.1 / Wavelength: 0.80000,0.91841 / Beamline: 14.1 / Wavelength: 0.80000,0.91841 | |||||||||
| Detector | Type: RAYONIX MX-225 / Detector: CCD / Date: Jun 5, 2009 / Details: mirrors | |||||||||
| Radiation | Monochromator: Double crystal / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||
| Radiation wavelength |
| |||||||||
| Reflection | Resolution: 0.98→20 Å / Num. all: 17179 / Num. obs: 17179 / % possible obs: 94.8 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Redundancy: 12.9 % / Biso Wilson estimate: 2.04 Å2 / Rmerge(I) obs: 0.072 / Net I/σ(I): 22.52 | |||||||||
| Reflection shell | Resolution: 0.98→1.01 Å / Redundancy: 2.3 % / Rmerge(I) obs: 0.46 / Mean I/σ(I) obs: 2.04 / Num. unique all: 1105 / % possible all: 84.5 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1AN1 Resolution: 0.98→20 Å / Num. parameters: 3821 / Num. restraintsaints: 3967 / Isotropic thermal model: ANISOTROPIC / Cross valid method: FREE R / σ(F): 0 / Stereochemistry target values: Engh & Huber Details: ANISOTROPIC REFINEMENT. CONJUGATE GRADIENT LEAST SQUARES REFINEMENT WITH RESTRAINTS APPLIED ONLY TO RESIDUES IN DOUBLE CONFORMATION. HYDROGEN ATOMS WERE ADDED AT RIDING POSITIONS
| |||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: MOEWS & KRETSINGER, J.MOL.BIOL.91(1973)201-228 | |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 17 / Occupancy sum hydrogen: 260.62 / Occupancy sum non hydrogen: 356.69 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 0.98→20 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
