Lysine-tRNA ligase, class II / Lysine-tRNA ligase, class II, N-terminal / Lysyl-tRNA synthetase, class II, C-terminal / Aminoacyl-tRNA synthetase, class II (D/K/N) / tRNA synthetases class II (D, K and N) / OB-fold nucleic acid binding domain, AA-tRNA synthetase-type / OB-fold nucleic acid binding domain / Bira Bifunctional Protein; Domain 2 / BirA Bifunctional Protein; domain 2 / Aminoacyl-tRNA synthetase, class II ...Lysine-tRNA ligase, class II / Lysine-tRNA ligase, class II, N-terminal / Lysyl-tRNA synthetase, class II, C-terminal / Aminoacyl-tRNA synthetase, class II (D/K/N) / tRNA synthetases class II (D, K and N) / OB-fold nucleic acid binding domain, AA-tRNA synthetase-type / OB-fold nucleic acid binding domain / Bira Bifunctional Protein; Domain 2 / BirA Bifunctional Protein; domain 2 / Aminoacyl-tRNA synthetase, class II / Aminoacyl-transfer RNA synthetases class-II family profile. / Class II Aminoacyl-tRNA synthetase/Biotinyl protein ligase (BPL) and lipoyl protein ligase (LPL) / Nucleic acid-binding proteins / OB fold (Dihydrolipoamide Acetyltransferase, E2P) / Nucleic acid-binding, OB-fold / Beta Barrel / 2-Layer Sandwich / Mainly Beta / Alpha Beta 類似検索 - ドメイン・相同性
温度: 289 K / 手法: 蒸気拡散法, シッティングドロップ法 / pH: 7.5 詳細: ButhA.00612.a.A1 PS01208 at 18.9 mg/mL against Morpheus H5, 10% PEG 20,000, 20% PEG 550 MME, 0.1 M MOPS/Hepes, 20 mM glutamate, 20 mM alanine, 20 mM glycine, 20 mM serine, 20 mM lysine, ...詳細: ButhA.00612.a.A1 PS01208 at 18.9 mg/mL against Morpheus H5, 10% PEG 20,000, 20% PEG 550 MME, 0.1 M MOPS/Hepes, 20 mM glutamate, 20 mM alanine, 20 mM glycine, 20 mM serine, 20 mM lysine, crystal tracking ID 232983h5, pH 7.5, VAPOR DIFFUSION, SITTING DROP, temperature 289K
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報 Burkholderia thailandensis (バクテリア)
Burkholderia thailandensis (バクテリア) X線回折 /
X線回折 /  データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード















 PDBj
PDBj

 集合体
集合体
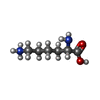
 タイプ: L-peptide linking / 分子量: 147.195 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C6H15N2O2
タイプ: L-peptide linking / 分子量: 147.195 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C6H15N2O2 分子量: 18.015 Da / 分子数: 461 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 461 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 解析
解析