 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
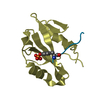
Yorodumi- PDB-4d6x: Crystal structure of the receiver domain of NtrX from Brucella abortus -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4d6x | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the receiver domain of NtrX from Brucella abortus | ||||||
 Components Components | BACTERIAL REGULATORY, FIS FAMILY PROTEIN | ||||||
 Keywords Keywords | SIGNALING PROTEIN / BRUCELLOSIS / TWO-COMPONENT SYSTEM / RESPONSE REGULATOR / REC DOMAIN / MICROAEROBISIS | ||||||
| Function / homology |  Function and homology information Function and homology informationphosphorelay signal transduction system / sequence-specific DNA binding / regulation of DNA-templated transcription / DNA-templated transcription / ATP binding / metal ion binding Similarity search - Function | ||||||
| Biological species |  BRUCELLA ABORTUS (bacteria) BRUCELLA ABORTUS (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.11 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.11 Å | ||||||
 Authors Authors | Klinke, S. / Fernandez, I. / Carrica, M.C. / Otero, L.H. / Goldbaum, F.A. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2015 Title: Snapshots of Conformational Changes Shed Light Into the Ntrx Receiver Domain Signal Transduction Mechanism Authors: Fernandez, I. / Otero, L.H. / Klinke, S. / Carrica, M.C. / Goldbaum, F.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4d6x.cif.gz | 200.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4d6x.ent.gz | 163.6 KB | Display | PDB format |
| PDBx/mmJSON format | 4d6x.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/d6/4d6xftp://data.pdbj.org/pub/pdb/validation_reports/d6/4d6x | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4d6yC  1l5zS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 16332.590 Da / Num. of mol.: 4 / Fragment: RECEIVER DOMAIN, RESIDUES 1-126 Source method: isolated from a genetically manipulated source Details: THE GENE SEQUENCE ENCODING THE RECEIVER DOMAIN C-TERMINAL REGION (RESIDUES 127-136) WAS OMITTED FROM THE FINAL CONSTRUCT DUE TO POTENTIAL CLASHES INTO THE CRYSTAL PACKING Source: (gene. exp.) BRUCELLA ABORTUS (bacteria) / Strain: BRUCELLA MELITENSIS BIOVAR ABORTUS 2308 / Plasmid: PET24A / Production host: #2: Chemical |   Mass: 69.085 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H5N2 Mass: 69.085 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H5N2#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 83 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 83 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.96 Å3/Da / Density % sol: 37.33 % / Description: NONE |
|---|---|
| Crystal grow | pH: 7 Details: PROTEIN WAS CRYSTALLIZED FROM 27% PEG 550 MME; 0.15 M MALIC ACID, PH 7.0; 0.1 M IMIDAZOLE, PH 6.9. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SOLEIL  / Beamline: PROXIMA 1 / Wavelength: 0.97857 / Beamline: PROXIMA 1 / Wavelength: 0.97857 |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Nov 23, 2013 / Details: KIRKPATRICK-BAEZ PAIR OF BI-MORPH MIRRORS |
| Radiation | Monochromator: CHANNEL CUT CRYOGENICALLY COOLED MONOCROMATOR CRYSTAL Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97857 Å / Relative weight: 1 |
| Reflection | Resolution: 2.11→40.64 Å / Num. obs: 30141 / % possible obs: 98.7 % / Observed criterion σ(I): 2 / Redundancy: 5.11 % / Biso Wilson estimate: 57.41 Å2 / Rmerge(I) obs: 0.05 / Net I/σ(I): 16.63 |
| Reflection shell | Resolution: 2.11→2.24 Å / Redundancy: 5.28 % / Rmerge(I) obs: 0.65 / Mean I/σ(I) obs: 1.98 / % possible all: 98.1 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1L5Z Resolution: 2.11→40.64 Å / Cor.coef. Fo:Fc: 0.9523 / Cor.coef. Fo:Fc free: 0.9399 / SU R Cruickshank DPI: 0.229 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.228 / SU Rfree Blow DPI: 0.163 / SU Rfree Cruickshank DPI: 0.165
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 74.48 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.398 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.11→40.64 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.11→2.18 Å / Total num. of bins used: 15
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
