 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4c02: Crystal structure of human ACVR1 (ALK2) in complex with FKBP12.6 ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4c02 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of human ACVR1 (ALK2) in complex with FKBP12.6 and dorsomorphin | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | TRANSFERASE/ISOMERASE / TRANSFERASE-ISOMERASE COMPLEX / TRANSFERASE / DORSOMORPHIN | |||||||||
| Function / homology |  Function and homology information Function and homology informationendocardial cushion cell fate commitment / mitral valve morphogenesis / BMP receptor complex / cardiac muscle cell fate commitment / atrial septum primum morphogenesis / endocardial cushion fusion / positive regulation of cardiac epithelial to mesenchymal transition / acute inflammatory response / positive regulation of determination of dorsal identity / smooth muscle cell differentiation ...endocardial cushion cell fate commitment / mitral valve morphogenesis / BMP receptor complex / cardiac muscle cell fate commitment / atrial septum primum morphogenesis / endocardial cushion fusion / positive regulation of cardiac epithelial to mesenchymal transition / acute inflammatory response / positive regulation of determination of dorsal identity / smooth muscle cell differentiation / activin receptor complex / endocardial cushion formation / transforming growth factor beta receptor activity, type I / activin receptor activity, type II / BMP receptor activity / activin receptor activity, type I / receptor protein serine/threonine kinase / transmembrane receptor protein serine/threonine kinase activity / transforming growth factor beta receptor activity, type II / pharyngeal system development / transforming growth factor beta receptor activity, type III / activin binding / cellular response to BMP stimulus / negative regulation of activin receptor signaling pathway / activin receptor signaling pathway / positive regulation of sequestering of calcium ion / cyclic nucleotide binding / negative regulation of calcium-mediated signaling / embryonic heart tube morphogenesis / negative regulation of insulin secretion involved in cellular response to glucose stimulus / gastrulation with mouth forming second / dorsal/ventral pattern formation / transforming growth factor beta binding / negative regulation of release of sequestered calcium ion into cytosol / neuronal action potential propagation / insulin secretion involved in cellular response to glucose stimulus / determination of left/right symmetry / neural crest cell migration / atrioventricular valve morphogenesis / response to redox state / 'de novo' protein folding / negative regulation of heart rate / branching involved in blood vessel morphogenesis / ventricular septum morphogenesis / FK506 binding / negative regulation of G1/S transition of mitotic cell cycle / positive regulation of axon regeneration / SMAD binding / germ cell development / positive regulation of intracellular signal transduction / peptide hormone binding / mesoderm formation / positive regulation of SMAD protein signal transduction / smooth muscle contraction / response to vitamin E / regulation of ossification / BMP signaling pathway / positive regulation of bone mineralization / regulation of cardiac muscle contraction by regulation of the release of sequestered calcium ion / positive regulation of osteoblast differentiation / negative regulation of signal transduction / calcium channel inhibitor activity / T cell proliferation / Ion homeostasis / regulation of release of sequestered calcium ion into cytosol by sarcoplasmic reticulum / sarcoplasmic reticulum membrane / release of sequestered calcium ion into cytosol / calcium channel regulator activity / calcium channel complex / transforming growth factor beta receptor signaling pathway / protein maturation / protein tyrosine kinase binding / peptidyl-prolyl cis-trans isomerase activity / RNA polymerase II CTD heptapeptide repeat P3 isomerase activity / RNA polymerase II CTD heptapeptide repeat P6 isomerase activity / negative regulation of extrinsic apoptotic signaling pathway / peptidylprolyl isomerase / calcium-mediated signaling / cellular response to growth factor stimulus / response to hydrogen peroxide / Stimuli-sensing channels / Z disc / osteoblast differentiation / apical part of cell / heart development / positive regulation of cytosolic calcium ion concentration / protein refolding / in utero embryonic development / transmembrane transporter binding / cell differentiation / protein kinase activity / positive regulation of cell migration / cadherin binding / signaling receptor binding / protein serine/threonine kinase activity / positive regulation of DNA-templated transcription / protein homodimerization activity / positive regulation of transcription by RNA polymerase II / ATP binding / metal ion binding Similarity search - Function | |||||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.17 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.17 Å | |||||||||
 Authors Authors | Williams, E. / Riesebos, E. / Vollmar, M. / Krojer, T. / Bradley, A. / Shrestha, L. / Kupinska, K. / von Delft, F. / Arrowsmith, C.H. / Edwards, A.M. ...Williams, E. / Riesebos, E. / Vollmar, M. / Krojer, T. / Bradley, A. / Shrestha, L. / Kupinska, K. / von Delft, F. / Arrowsmith, C.H. / Edwards, A.M. / Bountra, C. / Bullock, A. | |||||||||
 Citation Citation | Journal: Ph D Thesis Title: Crystal Structure of Human Acvr1 (Alk2) in Complex with Fkbp12.6 And Dorsomorphin Authors: Williams, E. / Riesebos, E. / Vollmar, M. / Krojer, T. / Bradley, A. / Shrestha, L. / Kupinska, K. / von Delft, F. / Arrowsmith, C.H. / Edwards, A.M. / Bountra, C. / Bullock, A. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4c02.cif.gz | 190.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4c02.ent.gz | 150.3 KB | Display | PDB format |
| PDBx/mmJSON format | 4c02.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 4c02_validation.pdf.gz | 715.6 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 4c02_full_validation.pdf.gz | 730.5 KB | Display | |
| Data in XML | 4c02_validation.xml.gz | 23.3 KB | Display | |
| Data in CIF | 4c02_validation.cif.gz | 32.2 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/c0/4c02ftp://data.pdbj.org/pub/pdb/validation_reports/c0/4c02 | HTTPS FTP |
-Related structure data












-Links
 PDBj
PDBj











- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 2 types, 2 molecules AB
| #1: Protein | Mass: 37398.746 Da / Num. of mol.: 1 / Fragment: KINASE DOMAIN, RESIDUES 172-499 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Plasmid: PFB-LIC-BSE / Production host:   SPODOPTERA FRUGIPERDA (fall armyworm) / Strain (production host): SF9 SPODOPTERA FRUGIPERDA (fall armyworm) / Strain (production host): SF9References: UniProt: Q04771, non-specific protein-tyrosine kinase, receptor protein serine/threonine kinase |
|---|---|
| #2: Protein | Mass: 11798.501 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Plasmid: PNIC28-BSA4 / Production host:  |
-Non-polymers , 4 types, 253 molecules 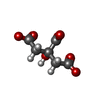



| #3: Chemical | ChemComp-FLC /  Mass: 189.100 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C6H5O7 Mass: 189.100 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C6H5O7#4: Chemical | ChemComp-TAK / |  Mass: 399.488 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C24H25N5O Mass: 399.488 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C24H25N5O#5: Chemical | ChemComp-EDO /  Mass: 62.068 Da / Num. of mol.: 13 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 13 / Source method: obtained synthetically / Formula: C2H6O2#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 231 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Sequence details | N TERMINAL METHIONINE |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 5.11 Å3/Da / Density % sol: 75.95 % / Description: NONE |
|---|---|
| Crystal grow | pH: 7.2 / Details: 1.8M AMMONIUM CITRATE, pH 7.2 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I04 / Wavelength: 0.9686 / Beamline: I04 / Wavelength: 0.9686 |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Jun 22, 2013 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9686 Å / Relative weight: 1 |
| Reflection | Resolution: 2.17→40 Å / Num. obs: 169113 / % possible obs: 99.9 % / Observed criterion σ(I): 2 / Redundancy: 9 % / Rmerge(I) obs: 0.17 / Net I/σ(I): 8 |
| Reflection shell | Resolution: 2.17→2.23 Å / Redundancy: 9.3 % / Rmerge(I) obs: 1.07 / Mean I/σ(I) obs: 2 / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRIES 1C9H AND 3H9R Resolution: 2.17→39.82 Å / Cor.coef. Fo:Fc: 0.957 / Cor.coef. Fo:Fc free: 0.949 / SU B: 6.385 / SU ML: 0.084 / Cross valid method: THROUGHOUT / ESU R: 0.125 / ESU R Free: 0.117 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS.U VALUES WITH TLS ADDED DISORDERED REGIONS WERE NOT MODELED AND RELEVANT LOOPS DELETED FROM STRUCTURE.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 36.836 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.17→39.82 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|