 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1c9h | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF FKBP12.6 IN COMPLEX WITH RAPAMYCIN | ||||||
 Components Components | FKBP12.6 | ||||||
 Keywords Keywords | IMMUNE SYSTEM / FKBP12 / RAPAMYCIN / COMPLEX / RYANODINE RECEPTOR | ||||||
| Function / homology |  Function and homology information Function and homology informationnegative regulation of calcium-mediated signaling / negative regulation of release of sequestered calcium ion into cytosol / response to redox state / negative regulation of heart rate / 'de novo' protein folding / FK506 binding / regulation of ryanodine-sensitive calcium-release channel activity / calcium channel inhibitor activity / regulation of release of sequestered calcium ion into cytosol by sarcoplasmic reticulum / Ion homeostasis ...negative regulation of calcium-mediated signaling / negative regulation of release of sequestered calcium ion into cytosol / response to redox state / negative regulation of heart rate / 'de novo' protein folding / FK506 binding / regulation of ryanodine-sensitive calcium-release channel activity / calcium channel inhibitor activity / regulation of release of sequestered calcium ion into cytosol by sarcoplasmic reticulum / Ion homeostasis / regulation of cardiac muscle contraction by regulation of the release of sequestered calcium ion / sarcoplasmic reticulum membrane / calcium channel complex / peptidylprolyl isomerase / peptidyl-prolyl cis-trans isomerase activity / calcium-mediated signaling / calcium channel regulator activity / protein maturation / Stimuli-sensing channels / Z disc / protein refolding / protein folding / transmembrane transporter binding / signaling receptor binding / membrane / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2 Å X-RAY DIFFRACTION / Resolution: 2 Å | ||||||
 Authors Authors | Deivanayagam, C.C.S. / Carson, M. / Thotakura, A. / Narayana, S.V.L. / Chodavarapu, C.S. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2000 Title: Structure of FKBP12.6 in complex with rapamycin. Authors: Deivanayagam, C.C. / Carson, M. / Thotakura, A. / Narayana, S.V. / Chodavarapu, R.S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1c9h.cif.gz | 37.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1c9h.ent.gz | 24.6 KB | Display | PDB format |
| PDBx/mmJSON format | 1c9h.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/c9/1c9hftp://data.pdbj.org/pub/pdb/validation_reports/c9/1c9h | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |












-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 11667.305 Da / Num. of mol.: 1 / Fragment: FKBP12.6 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CDNA / Organ: BRAIN / Plasmid: PET23A / Production host:  |
|---|---|
| #2: Chemical | ChemComp-RAP / 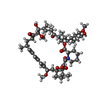  Mass: 914.172 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C51H79NO13 / Comment: antibiotic*YM Mass: 914.172 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C51H79NO13 / Comment: antibiotic*YM |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 101 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 101 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.49 Å3/Da / Density % sol: 50.67 % | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 7.4 Details: PEG 4000, LITHIUM SULFATE, HEPES BUFFER, pH 7.4, VAPOR DIFFUSION, HANGING DROP, temperature 298K | ||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS | ||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH3R / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE / Date: May 15, 1998 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2→100 Å / Num. all: 105747 / Num. obs: 7941 / % possible obs: 94.2 % / Observed criterion σ(I): 1 / Redundancy: 8.5 % / Biso Wilson estimate: 22.7 Å2 / Rmerge(I) obs: 0.065 / Net I/σ(I): 17.8 |
| Reflection shell | Resolution: 1.99→2.06 Å / Redundancy: 9.9 % / Rmerge(I) obs: 0.247 / % possible all: 86.6 |
| Reflection shell | *PLUS % possible obs: 86.6 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2→20 Å / Rfactor Rfree error: 0.01 / Data cutoff high absF: 783518.27 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 45.39 Å2 / ksol: 0.362 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 29 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.13 Å / Rfactor Rfree error: 0.031 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: 'CNS 0.4, O, OOPS' / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
