 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
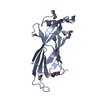
Yorodumi- PDB-3v31: Crystal Structure of the Peptide Bound Complex of the Ankyrin Rep... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3v31 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of the Peptide Bound Complex of the Ankyrin Repeat Domains of Human ANKRA2 | ||||||
 Components Components |
| ||||||
 Keywords Keywords | PROTEIN BINDING / Structural Genomics Consortium / SGC / ANKRA2 / ANK repeat / HDAC4 | ||||||
| Function / homology |  Function and homology information Function and homology informationnegative regulation of protein refolding / RUNX2 regulates chondrocyte maturation / response to denervation involved in regulation of muscle adaptation / negative regulation of myotube differentiation / positive regulation of protein sumoylation / : / cardiac muscle hypertrophy in response to stress / histone deacetylase activity, hydrolytic mechanism / histone deacetylase / SUMO transferase activity ...negative regulation of protein refolding / RUNX2 regulates chondrocyte maturation / response to denervation involved in regulation of muscle adaptation / negative regulation of myotube differentiation / positive regulation of protein sumoylation / : / cardiac muscle hypertrophy in response to stress / histone deacetylase activity, hydrolytic mechanism / histone deacetylase / SUMO transferase activity / histone deacetylase activity / protein lysine deacetylase activity / negative regulation of glycolytic process / low-density lipoprotein particle receptor binding / Notch-HLH transcription pathway / DNA-binding transcription activator activity / type I interferon-mediated signaling pathway / negative regulation of gene expression, epigenetic / histone deacetylase complex / potassium ion binding / B cell activation / RUNX3 regulates p14-ARF / protein sumoylation / regulation of protein-containing complex assembly / transcription repressor complex / B cell differentiation / epigenetic regulation of gene expression / response to interleukin-1 / SUMOylation of chromatin organization proteins / SUMOylation of intracellular receptors / NOTCH1 Intracellular Domain Regulates Transcription / Constitutive Signaling by NOTCH1 PEST Domain Mutants / Constitutive Signaling by NOTCH1 HD+PEST Domain Mutants / histone deacetylase binding / nervous system development / regulation of gene expression / DNA-binding transcription factor binding / molecular adaptor activity / RNA polymerase II-specific DNA-binding transcription factor binding / cytoskeleton / nuclear speck / RNA polymerase II cis-regulatory region sequence-specific DNA binding / chromatin remodeling / inflammatory response / positive regulation of cell population proliferation / ubiquitin protein ligase binding / protein kinase binding / positive regulation of DNA-templated transcription / chromatin / negative regulation of transcription by RNA polymerase II / DNA-templated transcription / positive regulation of transcription by RNA polymerase II / protein-containing complex / zinc ion binding / nucleoplasm / membrane / identical protein binding / nucleus / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.57 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.57 Å | ||||||
 Authors Authors | Lam, R. / Xu, C. / Bian, C.B. / Kania, J. / Bountra, C. / Weigelt, J. / Arrowsmith, C.H. / Edwards, A.M. / Bochkarev, A. / Min, J. / Structural Genomics Consortium (SGC) | ||||||
 Citation Citation | Journal: Sci.Signal. / Year: 2012 Title: Sequence-Specific Recognition of a PxLPxI/L Motif by an Ankyrin Repeat Tumbler Lock. Authors: Xu, C. / Jin, J. / Bian, C. / Lam, R. / Tian, R. / Weist, R. / You, L. / Nie, J. / Bochkarev, A. / Tempel, W. / Tan, C.S. / Wasney, G.A. / Vedadi, M. / Gish, G.D. / Arrowsmith, C.H. / ...Authors: Xu, C. / Jin, J. / Bian, C. / Lam, R. / Tian, R. / Weist, R. / You, L. / Nie, J. / Bochkarev, A. / Tempel, W. / Tan, C.S. / Wasney, G.A. / Vedadi, M. / Gish, G.D. / Arrowsmith, C.H. / Pawson, T. / Yang, X.J. / Min, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3v31.cif.gz | 84.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3v31.ent.gz | 62.6 KB | Display | PDB format |
| PDBx/mmJSON format | 3v31.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/v3/3v31ftp://data.pdbj.org/pub/pdb/validation_reports/v3/3v31 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3so8SC  3uxgC  3uzdC  3v2oC  3v2xC  3v30C S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 18171.551 Da / Num. of mol.: 1 / Fragment: UNP residues 148-313 (ANK repeats) Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: ANKRA, ANKRA2 / Plasmid: pET28-MHL / Production host:  | ||||
|---|---|---|---|---|---|
| #2: Protein/peptide | Mass: 1822.151 Da / Num. of mol.: 1 / Source method: obtained synthetically / Details: This sequence occurs naturally in human HDAC4. / Source: (synth.) Homo sapiens (human) / References: UniProt: P56524, histone deacetylase | ||||
| #3: Chemical | ChemComp-CL /   Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl | ||||
| #4: Chemical |   Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 139 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 139 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2 Å3/Da / Density % sol: 38.51 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: 0.1M Bis-Tris, pH 6.5, 0.2M NaCl, 25% PEG3350, VAPOR DIFFUSION, HANGING DROP, temperature 291K |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 19-ID / Wavelength: 0.97931 Å / Beamline: 19-ID / Wavelength: 0.97931 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Apr 14, 2010 Details: Rosenbaum-Rock high-resolution double-crystal monochromator | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: double-crystal monochromator / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.97931 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.57→50 Å / Num. obs: 22107 / % possible obs: 99.8 % / Redundancy: 4.4 % / Rmerge(I) obs: 0.041 / Χ2: 0.988 / Net I/σ(I): 16.8 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
-Phasing
| Phasing | Method: molecular replacement | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MR |
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 3SO8 Resolution: 1.57→26.41 Å / Cor.coef. Fo:Fc: 0.959 / Cor.coef. Fo:Fc free: 0.938 / WRfactor Rfree: 0.216 / WRfactor Rwork: 0.184 / Occupancy max: 1 / Occupancy min: 0.5 / SU B: 2.879 / SU ML: 0.049 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.09 / ESU R Free: 0.088 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : RESIDUAL ONLY
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 35.37 Å2 / Biso mean: 15.655 Å2 / Biso min: 4.43 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.57→26.41 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
