 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1sl6: Crystal Structure of a fragment of DC-SIGNR (containg the carbohy... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1sl6 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
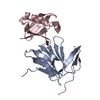
| Title | Crystal Structure of a fragment of DC-SIGNR (containg the carbohydrate recognition domain and two repeats of the neck) complexed with Lewis-x. | |||||||||
 Components Components | C-type lectin DC-SIGNR | |||||||||
 Keywords Keywords | SUGAR BINDING PROTEIN / DC-SIGNR / C-TYPE LECTIN | |||||||||
| Function / homology |  Function and homology information Function and homology informationcell-cell recognition / intracellular transport of virus / peptide antigen transport / ICAM-3 receptor activity / virion binding / antigen processing and presentation / leukocyte cell-cell adhesion / pattern recognition receptor activity / RSV-host interactions / D-mannose binding ...cell-cell recognition / intracellular transport of virus / peptide antigen transport / ICAM-3 receptor activity / virion binding / antigen processing and presentation / leukocyte cell-cell adhesion / pattern recognition receptor activity / RSV-host interactions / D-mannose binding / receptor-mediated endocytosis of virus by host cell / viral genome replication / peptide antigen binding / calcium-dependent protein binding / host cell / carbohydrate binding / virus receptor activity / signaling receptor activity / adaptive immune response / receptor-mediated virion attachment to host cell / intracellular signal transduction / immune response / external side of plasma membrane / innate immune response / symbiont entry into host cell / virion attachment to host cell / extracellular region / membrane / metal ion binding / plasma membrane / cytoplasm Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.25 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.25 Å | |||||||||
 Authors Authors | Guo, Y. / Feinberg, H. / Conroy, E. / Mitchell, D.A. / Alvarez, R. / Blixt, O. / Taylor, M.E. / Weis, W.I. / Drickamer, K. | |||||||||
 Citation Citation | Journal: Nat.Struct.Mol.Biol. / Year: 2004 Title: Structural basis for distinct ligand-binding and targeting properties of the receptors DC-SIGN and DC-SIGNR Authors: Guo, Y. / Feinberg, H. / Conroy, E. / Mitchell, D.A. / Alvarez, R. / Blixt, O. / Taylor, M.E. / Weis, W.I. / Drickamer, K. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1sl6.cif.gz | 232.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1sl6.ent.gz | 185.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1sl6.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/sl/1sl6ftp://data.pdbj.org/pub/pdb/validation_reports/sl/1sl6 | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| 5 | 
| ||||||||
| 6 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 21404.506 Da / Num. of mol.: 6 Source method: isolated from a genetically manipulated source Details: A fragment containing the CRD domain and two repeats from the neck domain Source: (gene. exp.) Homo sapiens (human) / Cell line (production host): BL21/DE3 / Production host:  #2: Polysaccharide | alpha-L-fucopyranose-(1-3)-[beta-D-galactopyranose-(1-4)]2-acetamido-2-deoxy-alpha-D-glucopyranose / Lewis X antigen / alpha anomer 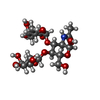  Source method: isolated from a genetically manipulated source Details: oligosaccharide with branches / References: Lewis X antigen, alpha anomer #3: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 24 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 24 / Source method: obtained synthetically / Formula: Ca#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 236 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 236 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.69 Å3/Da / Density % sol: 66.37 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: 30% PEG 300, 0.2M NaCl, 0.1 Hepes pH=7.5. Protein solution: 10 mg/ml protein, 5mM CaCl2, 10 mM oligosaccharide., VAPOR DIFFUSION, HANGING DROP, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS  / Beamline: 8.2.2 / Wavelength: 1.0781 Å / Beamline: 8.2.2 / Wavelength: 1.0781 Å |
| Detector | Type: ADSC / Detector: CCD / Date: Jul 3, 2003 |
| Radiation | Monochromator: Double-crystal Si(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.0781 Å / Relative weight: 1 |
| Reflection | Resolution: 2.25→50 Å / Num. all: 83241 / Num. obs: 80306 / Observed criterion σ(I): -3 / Biso Wilson estimate: 25.9 Å2 / Rsym value: 0.054 |
| Reflection shell | Resolution: 2.25→2.33 Å / Rsym value: 0.322 / % possible all: 84.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.25→41.96 Å / Rfactor Rfree error: 0.003 / Data cutoff high absF: 3723247.21 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 38.8621 Å2 / ksol: 0.348332 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 54.2 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.25→41.96 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.25→2.39 Å / Rfactor Rfree error: 0.013 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
