 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3rit: Crystal structure of Dipeptide Epimerase from Methylococcus capsu... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3rit | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Dipeptide Epimerase from Methylococcus capsulatus complexed with Mg and dipeptide L-Arg-D-Lys | ||||||
 Components Components | Dipeptide epimerase | ||||||
 Keywords Keywords | ISOMERASE / TIM barrel / chloromuconate cycloisomerase | ||||||
| Function / homology |  Function and homology information Function and homology informationracemase and epimerase activity, acting on amino acids and derivatives / Isomerases; Racemases and epimerases; Acting on amino acids and derivatives / racemase and epimerase activity / amino acid catabolic process / peptide metabolic process / magnesium ion binding Similarity search - Function | ||||||
| Biological species |  Methylococcus capsulatus (bacteria) Methylococcus capsulatus (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.701 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.701 Å | ||||||
 Authors Authors | Lukk, T. / Sakai, A. / Song, L. / Gerlt, J.A. / Nair, S.K. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2012 Title: Homology models guide discovery of diverse enzyme specificities among dipeptide epimerases in the enolase superfamily. Authors: Lukk, T. / Sakai, A. / Kalyanaraman, C. / Brown, S.D. / Imker, H.J. / Song, L. / Fedorov, A.A. / Fedorov, E.V. / Toro, R. / Hillerich, B. / Seidel, R. / Patskovsky, Y. / Vetting, M.W. / ...Authors: Lukk, T. / Sakai, A. / Kalyanaraman, C. / Brown, S.D. / Imker, H.J. / Song, L. / Fedorov, A.A. / Fedorov, E.V. / Toro, R. / Hillerich, B. / Seidel, R. / Patskovsky, Y. / Vetting, M.W. / Nair, S.K. / Babbitt, P.C. / Almo, S.C. / Gerlt, J.A. / Jacobson, M.P. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3rit.cif.gz | 361.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3rit.ent.gz | 294 KB | Display | PDB format |
| PDBx/mmJSON format | 3rit.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ri/3ritftp://data.pdbj.org/pub/pdb/validation_reports/ri/3rit | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3ijiC  3ijlC  3ijqC  3ik4C  3jvaC  3jw7C  3jzuC  3k1gC  3kumC  3q45C  3q4dC  3r0kC  3r0uC  3r10C  3r11C  3r1zC  3ro6C C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||||||||||||||||||||
| 2 | 
| |||||||||||||||||||||||||||||||||
| 3 | 
| |||||||||||||||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||||||||||||||
| Components on special symmetry positions |
| |||||||||||||||||||||||||||||||||
| Details | THE AUTHOR STATES THAT THE BIOLOGICAL UNIT OF THIS PROTEIN IS UNKNOWN. |
-Components
-Protein , 1 types, 5 molecules ABCDE
| #1: Protein | Mass: 39092.113 Da / Num. of mol.: 5 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Methylococcus capsulatus (bacteria) / Strain: Bath / Gene: MCA1834 / Plasmid: pET-17b / Production host: |
|---|
-Non-polymers , 6 types, 599 molecules 
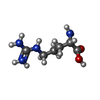
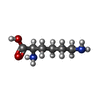



| #2: Chemical | ChemComp-SO4 /  Mass: 96.063 Da / Num. of mol.: 25 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 25 / Source method: obtained synthetically / Formula: SO4#3: Chemical | ChemComp-ARG /  Type: L-peptide linking / Mass: 175.209 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C6H15N4O2 Type: L-peptide linking / Mass: 175.209 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C6H15N4O2#4: Chemical | ChemComp-DLY /  Type: D-peptide linking / Mass: 146.188 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C6H14N2O2 Type: D-peptide linking / Mass: 146.188 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C6H14N2O2#5: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: Mg#6: Chemical | ChemComp-1PE /  Mass: 238.278 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H22O6 / Comment: precipitant*YM Mass: 238.278 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H22O6 / Comment: precipitant*YM#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 555 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.45 Å3/Da / Density % sol: 72.35 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 8.5 Details: Precipitant contained 25% PEG 3350, 0.1M Tris-HCl, and 0.2M Li2SO4. Protein solution contained 0.1M KCl, 0.05M HEPES (pH 8.0), 0.02M L-Arg-L-Lys, and 0.01M MgCl2. Protein concentration was ...Details: Precipitant contained 25% PEG 3350, 0.1M Tris-HCl, and 0.2M Li2SO4. Protein solution contained 0.1M KCl, 0.05M HEPES (pH 8.0), 0.02M L-Arg-L-Lys, and 0.01M MgCl2. Protein concentration was 12 mg/mL, pH 8.5, VAPOR DIFFUSION, HANGING DROP, temperature 277K |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 21-ID-G / Wavelength: 0.9786 Å / Beamline: 21-ID-G / Wavelength: 0.9786 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: RAYONIX MX-300 / Detector: CCD / Date: Mar 18, 2010 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: C(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.9786 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.7→19.87 Å / Num. all: 94589 / Num. obs: 94319 / % possible obs: 99.7 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 21.5 % / Biso Wilson estimate: 52.4 Å2 / Rmerge(I) obs: 0.154 / Net I/σ(I): 12.84 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.701→19.87 Å / SU ML: 0.77 / σ(F): 1.99 / Phase error: 21.86 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.83 Å / VDW probe radii: 1.1 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 19.947 Å2 / ksol: 0.31 e/Å3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.701→19.87 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
