 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3kgr: Crystal structure of the human leukocyte-associated Ig-like recep... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3kgr | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the human leukocyte-associated Ig-like receptor-1 (LAIR-1) | ||||||
 Components Components | Leukocyte-associated immunoglobulin-like receptor 1 | ||||||
 Keywords Keywords | IMMUNE SYSTEM / Ig-like domain / Cell membrane / Glycoprotein / Immune response / Immunoglobulin domain / Phosphoprotein / Receptor | ||||||
| Function / homology |  Function and homology information Function and homology informationimmune response-regulating signaling pathway / tertiary granule membrane / specific granule membrane / Immunoregulatory interactions between a Lymphoid and a non-Lymphoid cell / adaptive immune response / Neutrophil degranulation / plasma membrane Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å | ||||||
 Authors Authors | Brondijk, T.H.C. / Huizinga, E.G. / Ballering, J. | ||||||
 Citation Citation | Journal: Blood / Year: 2010 Title: Crystal structure and collagen-binding site of immune inhibitory receptor LAIR-1: unexpected implications for collagen binding by platelet receptor GPVI Authors: Brondijk, T.H.C. / de Ruiter, T. / Ballering, J. / Wienk, H. / Lebbink, R.J. / van Ingen, H. / Boelens, R. / Farndale, R.W. / Meyaard, L. / Huizinga, E.G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3kgr.cif.gz | 139.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3kgr.ent.gz | 110.3 KB | Display | PDB format |
| PDBx/mmJSON format | 3kgr.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/kg/3kgrftp://data.pdbj.org/pub/pdb/validation_reports/kg/3kgr | HTTPS FTP |
|---|
-Related structure data








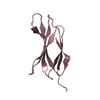




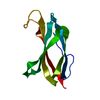
-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 11736.834 Da / Num. of mol.: 3 / Fragment: ectodomain Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: LAIR1 / Plasmid: pET3xa-hLAIR1 / Production host:  #2: Chemical | ChemComp-GLY / | 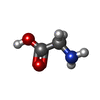  Type: peptide linking / Mass: 75.067 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H5NO2 Type: peptide linking / Mass: 75.067 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H5NO2#3: Chemical | ChemComp-GOL / |   Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 443 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 443 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.14 Å3/Da / Density % sol: 42.58 % / Mosaicity: 0.25 ° |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, sitting drop / pH: 4.3 Details: 25-30% PEG-1500, 100mM succinic acid, 100mM sodium dihydrogen phosphate, 100mM glycine, 10mM CoCl2, pH 4.3, VAPOR DIFFUSION, SITTING DROP, temperature 291K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID23-1 / Wavelength: 1.0396 Å / Beamline: ID23-1 / Wavelength: 1.0396 Å |
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Oct 7, 2007 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.0396 Å / Relative weight: 1 |
| Reflection twin | Operator: h,-h-k,-l / Fraction: 0.294 |
| Reflection | Resolution: 1.8→60.098 Å / Num. obs: 26097 / % possible obs: 96.3 % / Observed criterion σ(I): -3.7 / Redundancy: 5.5 % / Biso Wilson estimate: 17.458 Å2 / Rmerge(I) obs: 0.067 / Rsym value: 0.067 / Net I/σ(I): 20.2 |
| Reflection shell | Resolution: 1.8→1.9 Å / Redundancy: 3.9 % / Rmerge(I) obs: 0.208 / Mean I/σ(I) obs: 3.5 / Num. measured all: 12152 / Num. unique all: 3098 / Rsym value: 0.208 / % possible all: 78.4 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: Model from Swiss-model based on alignment with PDB entries 2GI7, 2D3V, 1UGN, 2GW5 Resolution: 1.8→34.698 Å / Occupancy max: 1 / Occupancy min: 0.29 / FOM work R set: 0.86 / Cross valid method: THROUGHOUT / σ(F): 2.05 / Phase error: 21.9 / Stereochemistry target values: TWIN_LSQ_F
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 59.86 Å2 / ksol: 0.361 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 99.03 Å2 / Biso mean: 23.233 Å2 / Biso min: 3.78 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→34.698 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 13 / % reflection obs: 98 %
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
