 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3bmc: Structure of Pteridine Reductase 1 (PTR1) from Trypanosoma brucei... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3bmc | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of Pteridine Reductase 1 (PTR1) from Trypanosoma brucei in ternary complex with cofactor (NADP+) and substrate (folate) | ||||||
 Components Components | (Pteridine reductase) x 2 | ||||||
 Keywords Keywords | OXIDOREDUCTASE / pteridine reductase / ptr1 / trypanosoma brucei / short chain dehydrogenase / substrate / folate | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.6 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.6 Å | ||||||
 Authors Authors | Tulloch, L.B. / Hunter, W.N. | ||||||
 Citation Citation | Journal: J.Med.Chem. / Year: 2010 Title: Structure-based design of pteridine reductase inhibitors targeting african sleeping sickness and the leishmaniases. Authors: Tulloch, L.B. / Martini, V.P. / Iulek, J. / Huggan, J.K. / Lee, J.H. / Gibson, C.L. / Smith, T.K. / Suckling, C.J. / Hunter, W.N. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3bmc.cif.gz | 207.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3bmc.ent.gz | 164.2 KB | Display | PDB format |
| PDBx/mmJSON format | 3bmc.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bm/3bmcftp://data.pdbj.org/pub/pdb/validation_reports/bm/3bmc | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3bmnC  3bmoC  3bmqC  3jq6C  3jq7C  3jq8C  3jq9C  3jqaC  3jqbC  3jqcC  3jqdC  3jqeC  3jqfC  3jqgC  3bms 3bmt C: citing same article ( |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: 1 / Ens-ID: 1 / Beg auth comp-ID: GLU / Beg label comp-ID: GLU / End auth comp-ID: FOL / End label comp-ID: FOL / Refine code: 1 / Auth seq-ID: 2 - 270 / Label seq-ID: 22
|
-Components
| #1: Protein | Mass: 30685.787 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #2: Protein | Mass: 30669.791 Da / Num. of mol.: 3 Source method: isolated from a genetically manipulated source Source: (gene. exp.) #3: Chemical | ChemComp-NAP /   Mass: 743.405 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C21H28N7O17P3 Mass: 743.405 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C21H28N7O17P3#4: Chemical | ChemComp-FOL / 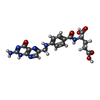  Mass: 441.397 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C19H19N7O6 Mass: 441.397 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C19H19N7O6#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 253 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 253 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.98 Å3/Da / Density % sol: 37.93 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 4.5 Details: 2-3M sodium acetate, 10-100mM sodium citrate, pH 4.5, vapor diffusion, hanging drop, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU MICROMAX-007 / Wavelength: 1.54 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE / Details: mirrors | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.54 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.6→73.737 Å / Num. obs: 23556 / % possible obs: 81.1 % / Redundancy: 2.2 % / Rmerge(I) obs: 0.068 / Rsym value: 0.068 / Net I/σ(I): 9.1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.6→37.67 Å / Cor.coef. Fo:Fc: 0.944 / Cor.coef. Fo:Fc free: 0.91 / SU B: 23.25 / SU ML: 0.249 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R Free: 0.421 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: All of the 17 related structures are expressed from the same DNA construct, which encodes CYS at positions 59 and 168. Crystals harvested within a couple of days of formation contain CYS at ...Details: All of the 17 related structures are expressed from the same DNA construct, which encodes CYS at positions 59 and 168. Crystals harvested within a couple of days of formation contain CYS at positions 59 and 168. However these two residues appear quite reactive and over time become oxidised to CSX, as determined by the emergence in older crystals of electron density for the OD atom. Sometimes CYS168 reacts with DTT in the crystallisation buffer, covalently linking the two molecules by an S-S bond.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 30.712 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.6→37.67 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Ens-ID: 1 / Number: 1784 / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.6→2.668 Å / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
