 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2ycw: TURKEY BETA1 ADRENERGIC RECEPTOR WITH STABILISING MUTATIONS AND B... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2ycw | ||||||
|---|---|---|---|---|---|---|---|
| Title | TURKEY BETA1 ADRENERGIC RECEPTOR WITH STABILISING MUTATIONS AND BOUND ANTAGONIST CARAZOLOL | ||||||
 Components Components | BETA-1 ADRENERGIC RECEPTOR | ||||||
 Keywords Keywords | RECEPTOR / GPCR / TRANSDUCER / ANTAGONIST BOUND FORM / INTEGRAL MEMBRANE PROTEIN / G-PROTEIN COUPLED RECEPTOR / THERMOSTABILISING POINT MUTATIONS / SEVEN-HELIX RECEPTOR / 7TM RECEPTOR | ||||||
| Function / homology |  Function and homology information Function and homology informationbeta1-adrenergic receptor activity / positive regulation of heart contraction / regulation of circadian sleep/wake cycle, sleep / adenylate cyclase-activating adrenergic receptor signaling pathway / early endosome / identical protein binding / membrane / plasma membrane Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3 Å | ||||||
 Authors Authors | Moukhametzianov, R. / Warne, T. / Edwards, P.C. / Serrano-Vega, M.J. / Leslie, A.G.W. / Tate, C.G. / Schertler, G.F.X. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2011 Title: Two Distinct Conformations of Helix 6 Observed in Antagonist-Bound Structures of a Beta-1- Adrenergic Receptor. Authors: Moukhametzianov, R. / Warne, T. / Edwards, P.C. / Serrano-Vega, M.J. / Leslie, A.G. / Tate, C.G. / Schertler, G.F. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2ycw.cif.gz | 135.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2ycw.ent.gz | 105.7 KB | Display | PDB format |
| PDBx/mmJSON format | 2ycw.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 2ycw_validation.pdf.gz | 1.8 MB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 2ycw_full_validation.pdf.gz | 1.8 MB | Display | |
| Data in XML | 2ycw_validation.xml.gz | 24.8 KB | Display | |
| Data in CIF | 2ycw_validation.cif.gz | 32.2 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/yc/2ycwftp://data.pdbj.org/pub/pdb/validation_reports/yc/2ycw | HTTPS FTP |
-Related structure data
| Related structure data |  2ycxC  2ycyC  2yczC  2vt4S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |















-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||||||||||||||||||||||||||
| 2 | 
| |||||||||||||||||||||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments:
|
-Components
| #1: Protein | Mass: 35752.598 Da / Num. of mol.: 2 / Fragment: RESIDUES 33-243,272-276,279-367 / Mutation: YES Source method: isolated from a genetically manipulated source Details: RESIDUES 3-32 AT THE N-TERMINUS AND RESIDUES 244-271 AND 277-278 OF THE THIRD INTRACELLULAR LOOP WERE DELETED FROM THE CONSTRUCT. THE CONSTRUCT WAS TRUNCATED AFTER RESIDUE 367 AND A HEXAHIS TAG ADDED. Source: (gene. exp.)  TRICHOPLUSIA NI (cabbage looper) / References: UniProt: P07700 TRICHOPLUSIA NI (cabbage looper) / References: UniProt: P07700#2: Chemical | 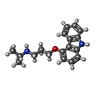  Mass: 298.379 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C18H22N2O2 Mass: 298.379 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C18H22N2O2#3: Chemical | ChemComp-2CV / 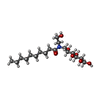  Mass: 379.489 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C18H37NO7 / Comment: detergent*YM Mass: 379.489 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C18H37NO7 / Comment: detergent*YM#4: Chemical |   Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: NaCompound details | ENGINEERED RESIDUE IN CHAIN A, ARG 68 TO SER ENGINEERED RESIDUE IN CHAIN A, MET 90 TO VAL ...ENGINEERED | Sequence details | THE FOLLOWING MUTATIONS WERE MADE TO IMPROVE THERMOSTABILITY R68S,M90V,Y227A,A282L,F327A,F338M THE ...THE FOLLOWING MUTATIONS WERE MADE TO IMPROVE THERMOSTAB | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.58 Å3/Da / Density % sol: 65.67 % Description: DATA WERE COLLECTED IN WEDGES, SCANNING THE MULTIPLE SPOTS ON THE CRYSTAL |
|---|---|
| Crystal grow | pH: 8.1 / Details: pH 8.1 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X06SA / Wavelength: 1 / Beamline: X06SA / Wavelength: 1 |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: May 1, 2008 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 3→44.7 Å / Num. obs: 17906 / % possible obs: 86 % / Observed criterion σ(I): 1.5 / Redundancy: 2.4 % / Biso Wilson estimate: 74.7 Å2 / Rmerge(I) obs: 0.1 / Net I/σ(I): 9.8 |
| Reflection shell | Resolution: 3→3.08 Å / Redundancy: 2 % / Rmerge(I) obs: 0.52 / Mean I/σ(I) obs: 1.6 / % possible all: 64 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2VT4 Resolution: 3→44.68 Å / Cor.coef. Fo:Fc: 0.91 / Cor.coef. Fo:Fc free: 0.86 / SU B: 19.335 / SU ML: 0.363 / Cross valid method: THROUGHOUT / ESU R Free: 0.483 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. U VALUES REFINED INDIVIDUALLY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 56.126 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3→44.68 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|