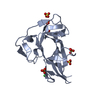
- PDB-2xvs: Crystal structure of human TTC5 (Strap) C-terminal OB domain -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 2xvs
Title
Crystal structure of human TTC5 (Strap) C-terminal OB domain
Components
TETRATRICOPEPTIDE REPEAT PROTEIN 5
Keywords
ANTITUMOR PROTEIN / P53 COFACTOR / STRESS-RESPONSE / P300
Function / homology
Function and homology information
positive regulation of mRNA catabolic process / cellular response to starvation / Regulation of TP53 Activity through Methylation / ribosome binding / cytoplasmic vesicle / mitochondrial matrix / DNA repair / DNA damage response / chromatin binding / positive regulation of transcription by RNA polymerase II ...positive regulation of mRNA catabolic process / cellular response to starvation / Regulation of TP53 Activity through Methylation / ribosome binding / cytoplasmic vesicle / mitochondrial matrix / DNA repair / DNA damage response / chromatin binding / positive regulation of transcription by RNA polymerase II / mitochondrion / DNA binding / nucleoplasm / cytosol Similarity search - Function
OB fold (Dihydrolipoamide Acetyltransferase, E2P) - #550 / Tetratricopeptide repeat protein 5, OB fold domain / TTC5, OB fold domain superfamily / Tetratricopeptide repeat protein 5 OB fold domain / TPR repeat region circular profile. / TPR repeat profile. / Tetratricopeptide repeats / Tetratricopeptide repeat / OB fold (Dihydrolipoamide Acetyltransferase, E2P) / Tetratricopeptide-like helical domain superfamily ...OB fold (Dihydrolipoamide Acetyltransferase, E2P) - #550 / Tetratricopeptide repeat protein 5, OB fold domain / TTC5, OB fold domain superfamily / Tetratricopeptide repeat protein 5 OB fold domain / TPR repeat region circular profile. / TPR repeat profile. / Tetratricopeptide repeats / Tetratricopeptide repeat / OB fold (Dihydrolipoamide Acetyltransferase, E2P) / Tetratricopeptide-like helical domain superfamily / Beta Barrel / Mainly Beta Similarity search - Domain/homology
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information HOMO SAPIENS (human)
HOMO SAPIENS (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads









 PDBj
PDBj


 Assembly
Assembly


 Mass: 126.904 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: I
Mass: 126.904 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: I
 Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2
 Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 18.015 Da / Num. of mol.: 116 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 116 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: X10SA / Wavelength: 0.99988
/ Beamline: X10SA / Wavelength: 0.99988  Processing
Processing