 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2wlk: STRUCTURE OF THE ATP-SENSITIVE INWARD RECTIFIER POTASSIUM CHANNEL... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2wlk | ||||||
|---|---|---|---|---|---|---|---|
| Title | STRUCTURE OF THE ATP-SENSITIVE INWARD RECTIFIER POTASSIUM CHANNEL FROM MAGNETOSPIRILLUM MAGNETOTACTICUM | ||||||
 Components Components | ATP-SENSITIVE INWARD RECTIFIER POTASSIUM CHANNEL 10 | ||||||
 Keywords Keywords | METAL TRANSPORT / INTEGRAL MEMBRANE PROTEIN / IONIC CHANNEL / ION TRANSPORT / TRANSPORT | ||||||
| Function / homology |  Function and homology information Function and homology informationinward rectifier potassium channel activity / regulation of monoatomic ion transmembrane transport / potassium ion import across plasma membrane / monoatomic ion channel complex / identical protein binding / plasma membrane Similarity search - Function | ||||||
| Biological species |  MAGNETOSPIRILLUM MAGNETOTACTICUM (bacteria) MAGNETOSPIRILLUM MAGNETOTACTICUM (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.8 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.8 Å | ||||||
 Authors Authors | Clarke, O.B. / Caputo, A.T. / Smith, B.J. / Gulbis, J.M. | ||||||
 Citation Citation | Journal: To be Published Title: Two Intermediate Gating State Crystal Structures of the Kirbac3.1 K+ Channel Authors: Gulbis, J.M. / Kuo, A. / Smith, B. / Doyle, D.A. / Edwards, A. / Arrowsmith, C. / Sundstrom, M. | ||||||
| History |
| ||||||
| Remark 0 | THIS ENTRY 2WLK REFLECTS AN ALTERNATIVE MODELING OF THE ORIGINAL STRUCTURAL DATA (R1XL6SF) ...THIS ENTRY 2WLK REFLECTS AN ALTERNATIVE MODELING OF THE ORIGINAL STRUCTURAL DATA (R1XL6SF) DETERMINED BY AUTHORS OF THE PDB ENTRY 1XL6: J.M.GULBIS,A.KUO,B.SMITH,D.A.DOYLE,A.EDWARDS,C.ARROWSMITH, M.SUNDSTROM | ||||||
| Remark 700 | SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN ... SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2wlk.cif.gz | 307.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2wlk.ent.gz | 259.4 KB | Display | PDB format |
| PDBx/mmJSON format | 2wlk.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/wl/2wlkftp://data.pdbj.org/pub/pdb/validation_reports/wl/2wlk | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2wlhC  2wliC  2wljC  2wllC  2wlmC  2wlnC  2wloC  2x6aC  2x6bC  2x6cC  1p7bS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components on special symmetry positions |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: 1 / Refine code: 1
NCS ensembles :
|
-Components
| #1: Protein | Mass: 33782.766 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) MAGNETOSPIRILLUM MAGNETOTACTICUM (bacteria)Plasmid: PET-30A / Production host: #2: Chemical | ChemComp-K /   Mass: 39.098 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: K Mass: 39.098 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: K#3: Chemical | ChemComp-CL / |   Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl#4: Chemical | ChemComp-SPM / | 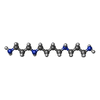  Mass: 202.340 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H26N4 Mass: 202.340 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H26N4#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 33 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 33 / Source method: isolated from a natural source / Formula: H2OSequence details | RESIDUES 5-295 CORRESPOND | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.77 Å3/Da / Density % sol: 65 % / Description: AUTHOR USED THE SF DATA FROM ENTRY 1XL6. |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 7.6 Details: 90MM HEPES, 2.5% PEG 8000, 2.5% PEG 4000, 20% PEG 400, 12.5MM MAGNESIUM CHLORIDE, 42.5MM MAGNESIUM ACETATE, 2.5% GLYCEROL, 14MM HEGA-10 , PH 7.6, VAPOR DIFFUSION, SITTING DROP, TEMPERATURE 293K |
-Data collection
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | Resolution: 2.7→30 Å / Num. obs: 28510 / Biso Wilson estimate: 0 Å2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1P7B Resolution: 2.8→28.76 Å / Cor.coef. Fo:Fc: 0.909 / Cor.coef. Fo:Fc free: 0.871 / SU B: 30.591 / SU ML: 0.284 / Cross valid method: THROUGHOUT / ESU R: 0 / ESU R Free: 0.402 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. ATOM RECORD CONTAINS RESIDUAL B FACTORS ONLY. REREFINEMENT OF PDB ENTRY 1XL6. IMPROVED REFINEMENT STATISTICS WERE OBTAINED, AND AN ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. ATOM RECORD CONTAINS RESIDUAL B FACTORS ONLY. REREFINEMENT OF PDB ENTRY 1XL6. IMPROVED REFINEMENT STATISTICS WERE OBTAINED, AND AN ALTERNATE CONFORMATION OF THE TRANSMEMBRANE DOMAIN WAS MODELLED.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 1 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 75.034 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.8→28.76 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|