 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2wgj: X-ray Structure of PF-02341066 bound to the kinase domain of c-Met -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2wgj | ||||||
|---|---|---|---|---|---|---|---|
| Title | X-ray Structure of PF-02341066 bound to the kinase domain of c-Met | ||||||
 Components Components | HEPATOCYTE GROWTH FACTOR RECEPTOR | ||||||
 Keywords Keywords | TRANSFERASE / C-MET / KINASE / INHIBITOR / ATP-BINDING / NUCLEOTIDE-BINDING / TYROSINE-PROTEIN KINASE | ||||||
| Function / homology |  Function and homology information Function and homology informationhepatocyte growth factor receptor activity / Drug-mediated inhibition of MET activation / MET activates STAT3 / negative regulation of hydrogen peroxide-mediated programmed cell death / MET Receptor Activation / MET interacts with TNS proteins / endothelial cell morphogenesis / semaphorin receptor activity / MET receptor recycling / pancreas development ...hepatocyte growth factor receptor activity / Drug-mediated inhibition of MET activation / MET activates STAT3 / negative regulation of hydrogen peroxide-mediated programmed cell death / MET Receptor Activation / MET interacts with TNS proteins / endothelial cell morphogenesis / semaphorin receptor activity / MET receptor recycling / pancreas development / MET activates PTPN11 / hepatocyte growth factor receptor signaling pathway / MET activates RAP1 and RAC1 / Sema4D mediated inhibition of cell attachment and migration / positive regulation of endothelial cell chemotaxis / MET activates PI3K/AKT signaling / MET activates PTK2 signaling / branching morphogenesis of an epithelial tube / positive chemotaxis / semaphorin-plexin signaling pathway / Regulation of MITF-M-dependent genes involved in cell cycle and proliferation / MET activates RAS signaling / MECP2 regulates neuronal receptors and channels / cell surface receptor protein tyrosine kinase signaling pathway / basal plasma membrane / molecular function activator activity / excitatory postsynaptic potential / liver development / negative regulation of autophagy / InlB-mediated entry of Listeria monocytogenes into host cell / receptor protein-tyrosine kinase / Negative regulation of MET activity / neuron differentiation / Constitutive Signaling by Aberrant PI3K in Cancer / PIP3 activates AKT signaling / PI5P, PP2A and IER3 Regulate PI3K/AKT Signaling / RAF/MAP kinase cascade / protein tyrosine kinase activity / protein phosphatase binding / cell surface receptor signaling pathway / postsynapse / signaling receptor complex / cell surface / positive regulation of transcription by RNA polymerase II / extracellular region / ATP binding / membrane / identical protein binding / plasma membrane Similarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | McTigue, M. / Grodsky, N. / Ryan, K. / Tran-Dube, M. / Cui, J.J. / Mroczkowski, B. | ||||||
 Citation Citation | Journal: J.Med.Chem / Year: 2011 Title: Structure Based Drug Design of Crizotinib (Pf-02341066), a Potent and Selective Dual Inhibitor of Mesenchymal-Epithelial Transition Factor (C-met) Kinase and Anaplastic Lymphoma Kinase (Alk). Authors: Cui, J.J. / Tran-Dube, M. / Shen, H. / Nambu, M. / Kung, P.P. / Pairish, M. / Jia, L. / Meng, J. / Funk, L. / Botrous, I. / Mctigue, M. / Grodsky, N. / Ryan, K. / Padrique, E. / Alton, G. / ...Authors: Cui, J.J. / Tran-Dube, M. / Shen, H. / Nambu, M. / Kung, P.P. / Pairish, M. / Jia, L. / Meng, J. / Funk, L. / Botrous, I. / Mctigue, M. / Grodsky, N. / Ryan, K. / Padrique, E. / Alton, G. / Timofeevski, S. / Yamazaki, S. / Li, Q. / Zou, H. / Christensen, J. / Mroczkowski, B. / Bender, S. / Kania, R.S. / Edwards, M.P. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2wgj.cif.gz | 78.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2wgj.ent.gz | 56.7 KB | Display | PDB format |
| PDBx/mmJSON format | 2wgj.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/wg/2wgjftp://data.pdbj.org/pub/pdb/validation_reports/wg/2wgj | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2wkmC  2xp2C  3lckS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |































-Links
 PDBj
PDBj










- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 34879.414 Da / Num. of mol.: 1 / Fragment: TYROSINE KINASE DOMAIN, RESIDUES 1051-1348 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Plasmid: PFASTBAC1 / Cell line (production host): SF9 / Production host:   SPODOPTERA FRUGIPERDA (fall armyworm) SPODOPTERA FRUGIPERDA (fall armyworm)References: UniProt: P08581, receptor protein-tyrosine kinase |
|---|---|
| #2: Chemical | ChemComp-VGH / 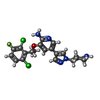  Mass: 450.337 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H22Cl2FN5O / Comment: medication*YM Mass: 450.337 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H22Cl2FN5O / Comment: medication*YM |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 180 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 180 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.36 Å3/Da / Density % sol: 47.5 % / Description: NONE |
|---|---|
| Crystal grow | Temperature: 286 K / Method: vapor diffusion, hanging drop / pH: 4.6 Details: CRYSTALS WERE GROWN BY HANGING DROP VAPOR DIFFUSION AT 13 DEGREES CELCIUS. 1-2 MICROLITERS OF PROTEIN SOLUTION AT 7-15 MG/ML WAS MIXEDWITH AN EQUAL VOLUME OF PRECIPITATING SOLUTION (0-275 MM ...Details: CRYSTALS WERE GROWN BY HANGING DROP VAPOR DIFFUSION AT 13 DEGREES CELCIUS. 1-2 MICROLITERS OF PROTEIN SOLUTION AT 7-15 MG/ML WAS MIXEDWITH AN EQUAL VOLUME OF PRECIPITATING SOLUTION (0-275 MM SODIUM CHLORIDE, 21% (W/V PEG 3350, 50 MM CITRATE-PHOSPHATE PH 4.6) |
-Data collection
| Diffraction | Mean temperature: 95 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS  / Beamline: 5.0.2 / Wavelength: 1 / Beamline: 5.0.2 / Wavelength: 1 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Apr 30, 2006 / Details: PINHOLE COLLIMATOR |
| Radiation | Monochromator: DOUBLE CRYSTAL SI(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2→50 Å / Num. obs: 18192 / % possible obs: 78.1 % / Observed criterion σ(I): 1 / Redundancy: 3.9 % / Rmerge(I) obs: 0.06 / Net I/σ(I): 20 |
| Reflection shell | Resolution: 2→2.05 Å / Redundancy: 2.8 % / Rmerge(I) obs: 0.29 / Mean I/σ(I) obs: 2.5 / % possible all: 38.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 3LCK Resolution: 2→19.96 Å / Cor.coef. Fo:Fc: 0.94 / Cor.coef. Fo:Fc free: 0.932 / SU B: 5.681 / SU ML: 0.145 / Cross valid method: THROUGHOUT / ESU R: 0.282 / ESU R Free: 0.196 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 40.859 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→19.96 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|