 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2vr3: Structural and Biochemical Characterization of Fibrinogen binding... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2vr3 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structural and Biochemical Characterization of Fibrinogen binding to ClfA from Staphylocccus aureus | ||||||
 Components Components |
| ||||||
 Keywords Keywords | CELL ADHESION / PLATELET AGGREGATION / PEPTIDOGLYCAN-ANCHOR / STAPHYLOCOCCUS AUREUS / FIBRINOGEN GAMMA-CHAIN / SECRETED / CELL WALL / VIRULENCE / CLUMPING FACTOR | ||||||
| Function / homology |  Function and homology information Function and homology informationaggregation of unicellular organisms / fibrinogen complex / platelet alpha granule / Regulation of TLR by endogenous ligand / fibrinogen binding / MyD88 deficiency (TLR2/4) / positive regulation of heterotypic cell-cell adhesion / IRAK4 deficiency (TLR2/4) / MyD88:MAL(TIRAP) cascade initiated on plasma membrane / plasminogen activation ...aggregation of unicellular organisms / fibrinogen complex / platelet alpha granule / Regulation of TLR by endogenous ligand / fibrinogen binding / MyD88 deficiency (TLR2/4) / positive regulation of heterotypic cell-cell adhesion / IRAK4 deficiency (TLR2/4) / MyD88:MAL(TIRAP) cascade initiated on plasma membrane / plasminogen activation / extracellular matrix structural constituent / p130Cas linkage to MAPK signaling for integrins / positive regulation of peptide hormone secretion / GRB2:SOS provides linkage to MAPK signaling for Integrins / fibronectin binding / positive regulation of exocytosis / protein secretion / blood coagulation, fibrin clot formation / protein polymerization / negative regulation of endothelial cell apoptotic process / positive regulation of vasoconstriction / Integrin cell surface interactions / : / negative regulation of extrinsic apoptotic signaling pathway via death domain receptors / fibrinolysis / Integrin signaling / positive regulation of substrate adhesion-dependent cell spreading / cell adhesion molecule binding / platelet alpha granule lumen / cell-matrix adhesion / positive regulation of protein secretion / Post-translational protein phosphorylation / Signaling by high-kinase activity BRAF mutants / MAP2K and MAPK activation / response to calcium ion / platelet aggregation / Regulation of Insulin-like Growth Factor (IGF) transport and uptake by Insulin-like Growth Factor Binding Proteins (IGFBPs) / Signaling by RAF1 mutants / Signaling by moderate kinase activity BRAF mutants / Paradoxical activation of RAF signaling by kinase inactive BRAF / Signaling downstream of RAS mutants / Signaling by BRAF and RAF1 fusions / Platelet degranulation / extracellular matrix / ER-Phagosome pathway / protein-containing complex assembly / blood microparticle / positive regulation of ERK1 and ERK2 cascade / cell adhesion / endoplasmic reticulum lumen / external side of plasma membrane / signaling receptor binding / structural molecule activity / cell surface / : / extracellular exosome / extracellular region / metal ion binding / plasma membrane Similarity search - Function | ||||||
| Biological species |   STAPHYLOCOCCUS AUREUS (bacteria) STAPHYLOCOCCUS AUREUS (bacteria) HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.95 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.95 Å | ||||||
 Authors Authors | Ganesh, V.K. / Rivera, J.J. / Smeds, E. / Bowden, M.G. / Wann, E.R. / Gurusidappa, S. / Fitzgerald, J.R. / Hook, M. | ||||||
 Citation Citation | Journal: Plos Pathog. / Year: 2008 Title: A Structural Model of the Staphylococcus Aureus Clfa-Fibrinogen Interaction Opens New Avenues for the Design of Anti-Staphylococcal Therapeutics. Authors: Ganesh, V.K. / Rivera, J.J. / Smeds, E. / Ko, Y.P. / Bowden, M.G. / Wann, E.R. / Gurusiddappa, S. / Fitzgerald, J.R. / Hook, M. | ||||||
| History |
| ||||||
| Remark 700 | SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN ... SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED. |
- Structure visualization
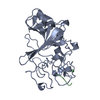
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2vr3.cif.gz | 143.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2vr3.ent.gz | 111.5 KB | Display | PDB format |
| PDBx/mmJSON format | 2vr3.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/vr/2vr3ftp://data.pdbj.org/pub/pdb/validation_reports/vr/2vr3 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1n67S S: Starting model for refinement |
|---|---|
| Similar structure data |






























-Links
 PDBj
PDBj










- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 35801.477 Da / Num. of mol.: 2 / Fragment: N2N3, RESIDUES 229-545 / Mutation: YES Source method: isolated from a genetically manipulated source Details: ENGINEERED DISULFIDE BOND BETWEEN 327 AND 541 / Source: (gene. exp.) STAPHYLOCOCCUS AUREUS (bacteria) / Production host: #2: Protein/peptide | Mass: 1276.424 Da / Num. of mol.: 2 Fragment: C-TERMINAL GAMMA-CHAIN PEPTIDE ANALOG, RESIDUES 425-437 Mutation: YES / Source method: obtained synthetically / Source: (synth.) HOMO SAPIENS (human) / References: UniProt: P02679#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 527 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 527 / Source method: isolated from a natural source / Formula: H2OCompound details | ENGINEERED RESIDUE IN CHAIN A, ASP 327 TO CYS ENGINEERED RESIDUE IN CHAIN A, LYS 541 TO CYS ...ENGINEERED | Has protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.3 Å3/Da / Density % sol: 47 % / Description: NONE |
|---|---|
| Crystal grow | pH: 6 / Details: 16-20% PEG 8000, 100MM SUCCINIC ACID PH 6.0 |
-Data collection
| Diffraction | Mean temperature: 120 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU / Wavelength: 1.5418 |
| Detector | Type: RIGAKU IMAGE PLATE / Detector: IMAGE PLATE / Details: MIRRORS |
| Radiation | Monochromator: GRAPHITE / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.95→15 Å / Num. obs: 49456 / % possible obs: 93.9 % / Observed criterion σ(I): 0 / Redundancy: 1.9 % / Rmerge(I) obs: 0.07 / Net I/σ(I): 5.8 |
| Reflection shell | Resolution: 1.95→2.02 Å / Redundancy: 1.75 % / Rmerge(I) obs: 0.198 / Mean I/σ(I) obs: 2.7 / % possible all: 88.7 |
- Processing
Processing
| Software | Name: REFMAC / Version: 5.2.0019 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1N67 Resolution: 1.95→15 Å / Cor.coef. Fo:Fc: 0.929 / Cor.coef. Fo:Fc free: 0.897 / SU B: 3.108 / SU ML: 0.092 / Cross valid method: THROUGHOUT / ESU R: 0.179 / ESU R Free: 0.191 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 29.927 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.95→15 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|