 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2fib: RECOMBINANT HUMAN GAMMA-FIBRINOGEN CARBOXYL TERMINAL FRAGMENT (RE... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2fib | ||||||
|---|---|---|---|---|---|---|---|
| Title | RECOMBINANT HUMAN GAMMA-FIBRINOGEN CARBOXYL TERMINAL FRAGMENT (RESIDUES 143-411) COMPLEXED TO THE PEPTIDE GLY-PRO-ARG-PRO AT PH 6.0 | ||||||
 Components Components |
| ||||||
 Keywords Keywords | COMPLEX (BLOOD COAGULATION/PEPTIDE) / FIBRINOGEN / BLOOD COAGULATION / FIBRIN POLYMERIZATION / COMPLEX (BLOOD COAGULATION-PEPTIDE) / COMPLEX (BLOOD COAGULATION-PEPTIDE) complex | ||||||
| Function / homology |  Function and homology information Function and homology informationfibrinogen complex / platelet alpha granule / Regulation of TLR by endogenous ligand / MyD88 deficiency (TLR2/4) / positive regulation of heterotypic cell-cell adhesion / IRAK4 deficiency (TLR2/4) / plasminogen activation / MyD88:MAL(TIRAP) cascade initiated on plasma membrane / extracellular matrix structural constituent / p130Cas linkage to MAPK signaling for integrins ...fibrinogen complex / platelet alpha granule / Regulation of TLR by endogenous ligand / MyD88 deficiency (TLR2/4) / positive regulation of heterotypic cell-cell adhesion / IRAK4 deficiency (TLR2/4) / plasminogen activation / MyD88:MAL(TIRAP) cascade initiated on plasma membrane / extracellular matrix structural constituent / p130Cas linkage to MAPK signaling for integrins / positive regulation of peptide hormone secretion / GRB2:SOS provides linkage to MAPK signaling for Integrins / positive regulation of exocytosis / blood coagulation, fibrin clot formation / protein polymerization / protein secretion / positive regulation of vasoconstriction / Integrin cell surface interactions / : / negative regulation of endothelial cell apoptotic process / negative regulation of extrinsic apoptotic signaling pathway via death domain receptors / fibrinolysis / Integrin signaling / positive regulation of substrate adhesion-dependent cell spreading / cell adhesion molecule binding / platelet alpha granule lumen / cell-matrix adhesion / positive regulation of protein secretion / Post-translational protein phosphorylation / Signaling by high-kinase activity BRAF mutants / MAP2K and MAPK activation / response to calcium ion / platelet aggregation / Regulation of Insulin-like Growth Factor (IGF) transport and uptake by Insulin-like Growth Factor Binding Proteins (IGFBPs) / Signaling by RAF1 mutants / Signaling by moderate kinase activity BRAF mutants / Paradoxical activation of RAF signaling by kinase inactive BRAF / Signaling downstream of RAS mutants / Signaling by BRAF and RAF1 fusions / Platelet degranulation / extracellular matrix / ER-Phagosome pathway / protein-containing complex assembly / blood microparticle / positive regulation of ERK1 and ERK2 cascade / endoplasmic reticulum lumen / signaling receptor binding / external side of plasma membrane / structural molecule activity / cell surface / : / extracellular exosome / extracellular region / metal ion binding / plasma membrane Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / ISOMORPHOUS WITH PDB ENTRY 1FIB / Resolution: 2.01 Å X-RAY DIFFRACTION / ISOMORPHOUS WITH PDB ENTRY 1FIB / Resolution: 2.01 Å | ||||||
 Authors Authors | Pratt, K.P. / Cote, H.C.F. / Chung, D.W. / Stenkamp, R.E. / Davie, E.W. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 1997 Title: The primary fibrin polymerization pocket: three-dimensional structure of a 30-kDa C-terminal gamma chain fragment complexed with the peptide Gly-Pro-Arg-Pro. Authors: Pratt, K.P. / Cote, H.C. / Chung, D.W. / Stenkamp, R.E. / Davie, E.W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2fib.cif.gz | 67.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2fib.ent.gz | 47.8 KB | Display | PDB format |
| PDBx/mmJSON format | 2fib.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fi/2fibftp://data.pdbj.org/pub/pdb/validation_reports/fi/2fib | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3fibC  1fibS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj










- Assembly
Assembly
| Deposited unit | 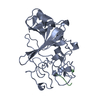
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 30243.422 Da / Num. of mol.: 1 Fragment: GAMMA CHAIN, CARBOXYL TERMINAL FRAGMENT RESIDUES 143 - 411 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Tissue: BLOOD PLASMAGene: HUMAN FIBRINOGEN GAMMA CHAIN CDNA ENCODING VAL 143 - VAL 411 Organ: BLOOD / Plasmid: PPIC9K / Production host:  Pichia pastoris (fungus) / References: UniProt: P02679 Pichia pastoris (fungus) / References: UniProt: P02679 |
|---|---|
| #2: Protein/peptide | Mass: 426.490 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source |
| #3: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 115 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 115 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
| Sequence details | NO DENSITY FOR RESIDUES BEYOND LEU 392 WAS OBSERVED. MASS SPECTROMET |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.91 Å3/Da / Density % sol: 35.52 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Method: vapor diffusion, sitting drop / pH: 6 Details: PROTEIN WAS CRYSTALLIZED FROM 12% PEG8000, 70MM CACL2, 0.1 M MES, PH 6.0, 0.02%NAN3, ROOM TEMPERATURE, SITTING DROPS. THE CRYSTAL WAS SOAKED OVERNIGHT IN THE ORIGINAL SOLUTION PLUS 0.01M GLY- ...Details: PROTEIN WAS CRYSTALLIZED FROM 12% PEG8000, 70MM CACL2, 0.1 M MES, PH 6.0, 0.02%NAN3, ROOM TEMPERATURE, SITTING DROPS. THE CRYSTAL WAS SOAKED OVERNIGHT IN THE ORIGINAL SOLUTION PLUS 0.01M GLY-PRO-ARG-PRO (SIGMA), THEN BACK-SOAKED FOR 10 MINUTES., vapor diffusion - sitting drop Temp details: room temp | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, sitting drop | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 300 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH2R / Wavelength: 1.5418 |
| Detector | Type: RIGAKU / Detector: IMAGE PLATE / Date: Sep 1, 1995 / Details: MIRRORS |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2→68.2 Å / Num. obs: 12275 / % possible obs: 76.9 % / Observed criterion σ(I): 3 / Rmerge(I) obs: 0.054 / Rsym value: 0.052 / Net I/σ(I): 11 |
| Reflection shell | Resolution: 2.01→2.25 Å / Mean I/σ(I) obs: 3.17 / % possible all: 61.3 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: ISOMORPHOUS WITH PDB ENTRY 1FIB Starting model: PDB ENTRY 1FIB Resolution: 2.01→10 Å / σ(F): 3 Details: MODIFIED TOPOLOGY AND PARAMETER FILES WERE CREATED SO THAT X-PLOR WOULD ACCEPT A CIS PEPTIDE BOND THAT DID NOT PRECEDE A PROLINE RESIDUE. THE PDB INPUT FILE FOR X-PLOR SHOULD SPECIFY CYS 339 ...Details: MODIFIED TOPOLOGY AND PARAMETER FILES WERE CREATED SO THAT X-PLOR WOULD ACCEPT A CIS PEPTIDE BOND THAT DID NOT PRECEDE A PROLINE RESIDUE. THE PDB INPUT FILE FOR X-PLOR SHOULD SPECIFY CYS 339 AS CCY 339. THE BACKBONE ANGLES PHI AND PSI OF ASN 337 ARE STRAINED.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 23.5 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.01→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.1→10 Å / Total num. of bins used: 20 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
