 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2vgq | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of Human IPS-1 CARD | |||||||||
 Components Components | Sugar ABC transporter substrate-binding protein,Mitochondrial antiviral-signaling protein | |||||||||
 Keywords Keywords | IMMUNE SYSTEM/TRANSPORT / IPS1/MAVS/VISA/CARDIF / CASPASE ACTIVATION / CASPASE RECRUITMENT DOMAIN / INNATE IMMUNITY / FUSION PROTEIN / SUGAR TRANSPORT / TRANSPORT / IMMUNE SYSTEM / CHIMERA / IMMUNE SYSTEM-TRANSPORT complex | |||||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of IP-10 production / regulation of peroxisome organization / positive regulation of chemokine (C-C motif) ligand 5 production / positive regulation of myeloid dendritic cell cytokine production / CARD domain binding / NF-kB activation through FADD/RIP-1 pathway mediated by caspase-8 and -10 / positive regulation of response to cytokine stimulus / protein localization to mitochondrion / positive regulation of type I interferon-mediated signaling pathway / peroxisomal membrane ...positive regulation of IP-10 production / regulation of peroxisome organization / positive regulation of chemokine (C-C motif) ligand 5 production / positive regulation of myeloid dendritic cell cytokine production / CARD domain binding / NF-kB activation through FADD/RIP-1 pathway mediated by caspase-8 and -10 / positive regulation of response to cytokine stimulus / protein localization to mitochondrion / positive regulation of type I interferon-mediated signaling pathway / peroxisomal membrane / TRAF6 mediated IRF7 activation / negative regulation of viral genome replication / cytoplasmic pattern recognition receptor signaling pathway / type I interferon-mediated signaling pathway / cellular response to exogenous dsRNA / detection of maltose stimulus / positive regulation of NLRP3 inflammasome complex assembly / maltose transport complex / carbohydrate transport / TRAF6 mediated NF-kB activation / positive regulation of interferon-alpha production / carbohydrate transmembrane transporter activity / maltose binding / positive regulation of type I interferon production / maltose transport / maltodextrin transmembrane transport / ubiquitin ligase complex / ATP-binding cassette (ABC) transporter complex, substrate-binding subunit-containing / positive regulation of defense response to virus by host / signaling adaptor activity / antiviral innate immune response / activation of innate immune response / ATP-binding cassette (ABC) transporter complex / Negative regulators of DDX58/IFIH1 signaling / protein serine/threonine kinase binding / positive regulation of interferon-beta production / positive regulation of interleukin-8 production / cell chemotaxis / DDX58/IFIH1-mediated induction of interferon-alpha/beta / Evasion by RSV of host interferon responses / molecular condensate scaffold activity / mitochondrial membrane / PKR-mediated signaling / positive regulation of interleukin-6 production / positive regulation of protein import into nucleus / SARS-CoV-1 activates/modulates innate immune responses / Ovarian tumor domain proteases / positive regulation of tumor necrosis factor production / outer membrane-bounded periplasmic space / TRAF3-dependent IRF activation pathway / protein-macromolecule adaptor activity / molecular adaptor activity / defense response to virus / DNA-binding transcription factor binding / mitochondrial outer membrane / periplasmic space / positive regulation of canonical NF-kappaB signal transduction / intracellular signal transduction / defense response to bacterium / innate immune response / DNA damage response / protein kinase binding / SARS-CoV-2 activates/modulates innate and adaptive immune responses / signal transduction / positive regulation of transcription by RNA polymerase II / mitochondrion / identical protein binding / membrane Similarity search - Function | |||||||||
| Biological species |   Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å | |||||||||
 Authors Authors | Potter, J.A. / Randall, R.E. / Taylor, G.L. | |||||||||
 Citation Citation | Journal: BMC Struct Biol / Year: 2008 Title: Crystal structure of human IPS-1/MAVS/VISA/Cardif caspase activation recruitment domain. Authors: Jane A Potter / Richard E Randall / Garry L Taylor /  Abstract: BACKGROUND: IPS-1/MAVS/VISA/Cardif is an adaptor protein that plays a crucial role in the induction of interferons in response to viral infection. In the initial stage of the intracellular antiviral ...BACKGROUND: IPS-1/MAVS/VISA/Cardif is an adaptor protein that plays a crucial role in the induction of interferons in response to viral infection. In the initial stage of the intracellular antiviral response two RNA helicases, retinoic acid inducible gene-I (RIG-I) and melanoma differentiation-association gene 5 (MDA5), are independently able to bind viral RNA in the cytoplasm. The 62 kDa protein IPS-1/MAVS/VISA/Cardif contains an N-terminal caspase activation and recruitment (CARD) domain that associates with the CARD regions of RIG-I and MDA5, ultimately leading to the induction of type I interferons. As a first step towards understanding the molecular basis of this important adaptor protein we have undertaken structural studies of the IPS-1 MAVS/VISA/Cardif CARD region. RESULTS: The crystal structure of human IPS-1/MAVS/VISA/Cardif CARD has been determined to 2.1A resolution. The protein was expressed and crystallized as a maltose-binding protein (MBP) fusion ...RESULTS: The crystal structure of human IPS-1/MAVS/VISA/Cardif CARD has been determined to 2.1A resolution. The protein was expressed and crystallized as a maltose-binding protein (MBP) fusion protein. The MBP and IPS-1 components each form a distinct domain within the structure. IPS-1/MAVS/VISA/Cardif CARD adopts a characteristic six-helix bundle with a Greek-key topology and, in common with a number of other known CARD structures, contains two major polar surfaces on opposite sides of the molecule. One face has a surface-exposed, disordered tryptophan residue that may explain the poor solubility of untagged expression constructs. CONCLUSION: The IPS-1/MAVS/VISA/Cardif CARD domain adopts the classic CARD fold with an asymmetric surface charge distribution that is typical of CARD domains involved in homotypic protein-protein ...CONCLUSION: The IPS-1/MAVS/VISA/Cardif CARD domain adopts the classic CARD fold with an asymmetric surface charge distribution that is typical of CARD domains involved in homotypic protein-protein interactions. The location of the two polar areas on IPS-1/MAVS/VISA/Cardif CARD suggest possible types of associations that this domain makes with the two CARD domains of MDA5 or RIG-I. The N-terminal CARD domains of RIG-I and MDA5 share greatest sequence similarity with IPS-1/MAVS/VISA/Cardif CARD and this has allowed modelling of their structures. These models show a very different charge profile for the equivalent surfaces compared to IPS-1/MAVS/VISA/Cardif CARD. | |||||||||
| History |
| |||||||||
| Remark 700 | SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN ... SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2vgq.cif.gz | 110.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2vgq.ent.gz | 84.1 KB | Display | PDB format |
| PDBx/mmJSON format | 2vgq.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 2vgq_validation.pdf.gz | 825.9 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 2vgq_full_validation.pdf.gz | 829.7 KB | Display | |
| Data in XML | 2vgq_validation.xml.gz | 20.1 KB | Display | |
| Data in CIF | 2vgq_validation.cif.gz | 29.5 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/vg/2vgqftp://data.pdbj.org/pub/pdb/validation_reports/vg/2vgq | HTTPS FTP |
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |






















































-Links
 PDBj
PDBj











- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 53411.324 Da / Num. of mol.: 1 Fragment: MMBP RESIDUES 27-392, CARD DOMAIN RESIDUES 3-93,MMBP RESIDUES 27-392, CARD DOMAIN RESIDUES 3-93 Source method: isolated from a genetically manipulated source Details: THE CONSTRUCT IS A FUSION OF E. COLI MBP (RESIDUES 2-366) AND HUMAN IPS-1 CARD (RESIDUES 1 TO 93) Source: (gene. exp.) Homo sapiens (human)Gene: malE, PU06_05845, MAVS, IPS1, KIAA1271, VISA / Production host: References: UniProt: A0A0B1N7A9, UniProt: Q7Z434, UniProt: P0AEX9*PLUS | ||
|---|---|---|---|
| #2: Polysaccharide | alpha-D-glucopyranose-(1-4)-alpha-D-glucopyranose-(1-4)-alpha-D-glucopyranose-(1-4)-alpha-D-glucopyranose / alpha-maltotetraose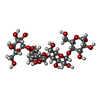  Source method: isolated from a genetically manipulated source Details: oligosaccharide / References: alpha-maltotetraose | ||
| #3: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: SO4#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 203 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 203 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.6 Å3/Da / Density % sol: 66 % / Description: NONE |
|---|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-2 / Wavelength: 0.934 / Beamline: ID14-2 / Wavelength: 0.934 |
| Detector | Type: ADSC CCD / Detector: CCD |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.934 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→32.7 Å / Num. obs: 48262 / % possible obs: 99.8 % / Observed criterion σ(I): 4.4 / Redundancy: 7.1 % / Rmerge(I) obs: 0.07 / Net I/σ(I): 16.1 |
| Reflection shell | Resolution: 2.1→2.21 Å / Redundancy: 7.2 % / Rmerge(I) obs: 0.36 / Mean I/σ(I) obs: 4.4 / % possible all: 99.9 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.1→32.24 Å / Cor.coef. Fo:Fc: 0.952 / Cor.coef. Fo:Fc free: 0.926 / SU B: 6.449 / SU ML: 0.09 / Cross valid method: THROUGHOUT / ESU R: 0.14 / ESU R Free: 0.138 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 21.55 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→32.24 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|