Entry Database : PDB / ID : 2vasTitle Myosin VI (MD-insert2-CaM, Delta-Insert1) Post-rigor state Keywords / / / / / / / / / / / / / / / / / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species SUS SCROFA (pig)DROSOPHILA MELANOGASTER (fruit fly)Method / / / Resolution : 2.4 Å Authors Menetrey, J. / Llinas, P. / Cicolari, J. / Squires, G. / Liu, X. / Li, A. / Sweeney, H.L. / Houdusse, A. Journal : Embo J. / Year : 2008Title : The Post-Rigor Structure of Myosin Vi and Implications for the Recovery Stroke.Authors : Menetrey, J. / Llinas, P. / Cicolari, J. / Squires, G. / Liu, X. / Li, A. / Sweeney, H.L. / Houdusse, A. History Deposition Sep 4, 2007 Deposition site / Processing site Revision 1.0 Dec 11, 2007 Provider / Type Revision 1.1 May 8, 2011 Group Revision 1.2 Jul 13, 2011 Group Revision 1.3 Dec 13, 2023 Group Data collection / Database references ... Data collection / Database references / Derived calculations / Other / Refinement description Category chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_database_status / pdbx_initial_refinement_model / pdbx_struct_conn_angle / struct_conn / struct_site Item _database_2.pdbx_DOI / _database_2.pdbx_database_accession ... _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_database_status.status_code_sf / _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_asym_id / _pdbx_struct_conn_angle.ptnr1_label_atom_id / _pdbx_struct_conn_angle.ptnr1_label_comp_id / _pdbx_struct_conn_angle.ptnr1_label_seq_id / _pdbx_struct_conn_angle.ptnr2_auth_comp_id / _pdbx_struct_conn_angle.ptnr2_auth_seq_id / _pdbx_struct_conn_angle.ptnr2_label_asym_id / _pdbx_struct_conn_angle.ptnr2_label_atom_id / _pdbx_struct_conn_angle.ptnr2_label_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_asym_id / _pdbx_struct_conn_angle.ptnr3_label_atom_id / _pdbx_struct_conn_angle.ptnr3_label_comp_id / _pdbx_struct_conn_angle.ptnr3_label_seq_id / _pdbx_struct_conn_angle.value / _struct_conn.pdbx_dist_value / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_conn.ptnr2_label_seq_id / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id
Show all Show less
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information

 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads













 PDBj
PDBj








 Assembly
Assembly
 SPODOPTERA FRUGIPERDA (fall armyworm) / References: UniProt: Q29122
SPODOPTERA FRUGIPERDA (fall armyworm) / References: UniProt: Q29122
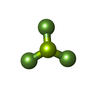



 Mass: 427.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM
Mass: 427.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Mass: 66.007 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: BeF3
Mass: 66.007 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: BeF3 Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca
Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca Sample preparation
Sample preparation / Beamline: ID23-2 / Wavelength: 0.8726
/ Beamline: ID23-2 / Wavelength: 0.8726  Processing
Processing