 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 2q4y | ||||||
|---|---|---|---|---|---|---|---|
| タイトル | Ensemble refinement of the protein crystal structure of At1g77540-coenzyme A complex | ||||||
 要素 要素 | Uncharacterized protein At1g77540 | ||||||
 キーワード キーワード | TRANSFERASE / Ensemble Refinement / Refinement Methodology Development / CoA / Coenzyme-A / COG2388 Family / acetyltransferase / At1g77540 / Structural Genomics / Protein Structure Initiative / PSI / Center for Eukaryotic Structural Genomics / CESG | ||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報 | ||||||
| 生物種 |  | ||||||
| 手法 |  X線回折 / Re-refinement using ensemble model / 解像度: 2.06 Å X線回折 / Re-refinement using ensemble model / 解像度: 2.06 Å | ||||||
 データ登録者 データ登録者 | Levin, E.J. / Kondrashov, D.A. / Wesenberg, G.E. / Phillips Jr., G.N. / Center for Eukaryotic Structural Genomics (CESG) | ||||||
 引用 引用 | ジャーナル: Structure / 年: 2007 タイトル: Ensemble refinement of protein crystal structures: validation and application. 著者: Levin, E.J. / Kondrashov, D.A. / Wesenberg, G.E. / Phillips, G.N. | ||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 2q4y.cif.gz | 92.6 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb2q4y.ent.gz | 73 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 2q4y.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| 文書・要旨 | 2q4y_validation.pdf.gz | 809.9 KB | 表示 | wwPDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | 2q4y_full_validation.pdf.gz | 865.3 KB | 表示 | |
| XML形式データ | 2q4y_validation.xml.gz | 30.7 KB | 表示 | |
| CIF形式データ | 2q4y_validation.cif.gz | 34.4 KB | 表示 | |
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/q4/2q4yftp://data.pdbj.org/pub/pdb/validation_reports/q4/2q4y | HTTPS FTP |
-関連構造データ
| 関連構造データ |  2q3mC  2q3oC  2q3pC  2q3qC  2q3rC  2q3sC  2q3tC  2q3uC  2q3vC  2q3wC  2q40C  2q41C  2q42C  2q43C  2q44C  2q45C  2q46C  2q47C  2q48C  2q49C  2q4aC  2q4bC  2q4cC  2q4dC  2q4eC  2q4fC  2q4hC  2q4iC  2q4jC  2q4kC  2q4lC  2q4mC  2q4nC  2q4oC  2q4pC  2q4qC  2q4rC  2q4sC  2q4tC  2q4uC  2q4vC  2q4xC  2q4zC  2q50C  2q51C  2q52C  2il4S S: 精密化の開始モデル C: 同じ文献を引用 ( |
|---|---|
| 類似構造データ | |
| その他のデータベース |











-リンク
 PDBj
PDBj





- 集合体
集合体
| 登録構造単位 | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| 単位格子 |
| ||||||||
| モデル数 | 4 |
-要素
| #1: タンパク質 | 分子量: 11752.433 Da / 分子数: 1 / 断片: Residues 12-114 / 由来タイプ: 組換発現 由来: (組換発現) 株: cv. Columbia / 遺伝子: At1g77540, T5M16.13 / プラスミド: PVP13-GW / 発現宿主:  |
|---|---|
| #2: 化合物 | ChemComp-COA / 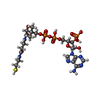  分子量: 767.534 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C21H36N7O16P3S 分子量: 767.534 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C21H36N7O16P3S |
| #3: 水 | ChemComp-HOH /  分子量: 18.015 Da / 分子数: 68 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 68 / 由来タイプ: 天然 / 式: H2O |
-実験情報
-実験
| 実験 | 手法: X線回折 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 2.24 Å3/Da / 溶媒含有率: 45.08 % / 解説: AUTHOR USED THE SF DATA FROM ENTRY 2IL4. |
|---|
-データ収集
| 放射 | プロトコル: SINGLE WAVELENGTH / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray |
|---|---|
| 放射波長 | 相対比: 1 |
- 解析
解析
| ソフトウェア |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: Re-refinement using ensemble model 開始モデル: PDB entry 2IL4 解像度: 2.06→31.97 Å / Rfactor Rfree error: 0.012 / Data cutoff high absF: 607463.438 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / 交差検証法: THROUGHOUT / σ(F): 0 立体化学のターゲット値: maximum likelihood using amplitudes 詳細: This PDB entry is a re-refinement using an ensemble model of the previously deposited single-conformer structure 2il4 and the first data set in the deposited structure factor file for 2il4 ...詳細: This PDB entry is a re-refinement using an ensemble model of the previously deposited single-conformer structure 2il4 and the first data set in the deposited structure factor file for 2il4 along with the R-free set defined therein. The coordinates were generated by an automated protocol from an initial model consisting of 4 identical copies of the protein and non-water hetero-atoms assigned fractional occupancies adding up to one, and a single copy of the solvent molecules. Refinement was carried out with all the conformers present simultaneously and with the potential energy terms corresponding to interactions between the different conformers excluded. The helix and sheet records were calculated using coordinates from the first conformer only. The structure visualization program PYMOL is well-suited for directly viewing the ensemble model presented in this PDB file.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | 溶媒モデル: FLAT MODEL / Bsol: 72.641 Å2 / ksol: 0.405 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | Biso mean: 29.6 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 2.06→31.97 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS精密化 シェル | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
