 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2bml: Ofloxacin-like antibiotics inhibit pneumococcal cell wall degradi... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2bml | ||||||
|---|---|---|---|---|---|---|---|
| Title | Ofloxacin-like antibiotics inhibit pneumococcal cell wall degrading virulence factors | ||||||
 Components Components | AUTOLYSIN | ||||||
 Keywords Keywords | CHOLINE-BINDING DOMAIN / CELL WALL ATTACHMENT / OFLOXACIN-LIKE ANTIBIOTICS | ||||||
| Function / homology |  Function and homology information Function and homology informationN-acetylmuramoyl-L-alanine amidase / establishment of competence for transformation / N-acetylmuramoyl-L-alanine amidase activity / sporulation resulting in formation of a cellular spore / peptidoglycan catabolic process / cell wall organization / extracellular region Similarity search - Function | ||||||
| Biological species |   STREPTOCOCCUS PNEUMONIAE (bacteria) STREPTOCOCCUS PNEUMONIAE (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.6 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.6 Å | ||||||
 Authors Authors | Fernandez-Tornero, C. / Gimenez-Gallego, G. / Romero, A. / Garcia, E. / Pascual-Teresa, B.D. / Lopez, R. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2005 Title: Ofloxacin-Like Antibiotics Inhibit Pneumococcal Cell Wall-Degrading Virulence Factors Authors: Fernandez-Tornero, C. / Garcia, E. / Lopez, R. / Pascual-Teresa, B.D. / Gimenez-Gallego, G. / Romero, A. #1: Journal: Nat.Struct.Biol. / Year: 2001Title: A Novel Solenoid Fold in the Cell Wall Anchoring Domain of the Pneumococcal Virulence Factor Lyta Authors: Fernandez-Tornero, C. / Garcia, E. / Lopez, R. / Gimenez-Gallego, G. / Romero, A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2bml.cif.gz | 69.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2bml.ent.gz | 51.3 KB | Display | PDB format |
| PDBx/mmJSON format | 2bml.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bm/2bmlftp://data.pdbj.org/pub/pdb/validation_reports/bm/2bml | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1hcxS S: Starting model for refinement |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (-0.95447, 0.29818, 0.00892), Vector: |
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 14892.502 Da / Num. of mol.: 2 / Fragment: CHOLINE-BINDING DOMAIN, RESIDUES 193-318 Source method: isolated from a genetically manipulated source Source: (gene. exp.) STREPTOCOCCUS PNEUMONIAE (bacteria) / Plasmid: PCE17 / Production host: References: UniProt: P06653, N-acetylmuramoyl-L-alanine amidase |
|---|
-Non-polymers , 6 types, 64 molecules 
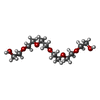
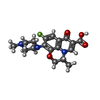



| #2: Chemical |  Mass: 122.143 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM Mass: 122.143 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM#3: Chemical |  Mass: 282.331 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H26O7 / Comment: precipitant*YM Mass: 282.331 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H26O7 / Comment: precipitant*YM#4: Chemical | ChemComp-XED / |  Mass: 361.368 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H20FN3O4 Mass: 361.368 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H20FN3O4#5: Chemical | ChemComp-PG4 / |  Mass: 194.226 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM Mass: 194.226 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM#6: Chemical |  Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 55 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.88 Å3/Da / Density % sol: 56.92 % |
|---|---|
| Crystal grow | pH: 7.5 Details: CRYSTALS WERE GROWN FROM A 12 MG/ML PROTEIN SOLUTION OVER A WELL SOLUTION CONTAINING 2M AMMONIUM SULPHATE AND 2% (W/V) PEG 400 BUFFERED WITH 0.1M HEPES (PH 7.5). |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: BM14 / Wavelength: 1.0163 / Beamline: BM14 / Wavelength: 1.0163 |
| Detector | Type: MARRESEARCH / Detector: CCD / Details: TOROIDAL MIRROR |
| Radiation | Monochromator: DOUBLE CRYSTAL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.0163 Å / Relative weight: 1 |
| Reflection | Resolution: 2.6→20 Å / Num. obs: 9193 / % possible obs: 87.1 % / Observed criterion σ(I): 0 / Redundancy: 4.6 % / Biso Wilson estimate: 49.8 Å2 / Rmerge(I) obs: 0.06 / Net I/σ(I): 8.6 |
| Reflection shell | Resolution: 2.6→20 Å / Redundancy: 4 % / Rmerge(I) obs: 0.27 / Mean I/σ(I) obs: 2.6 / % possible all: 91.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1HCX Resolution: 2.6→6 Å / Rfactor Rfree error: 0.01 / Data cutoff high absF: 704892.21 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: MAXIMUM LIKELIHOOD
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 82.541 Å2 / ksol: 0.676592 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 46.6 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.6→6 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.6→2.75 Å / Rfactor Rfree error: 0.035 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
