 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1gvm: CHOLINE BINDING DOMAIN OF THE MAJOR AUTOLYSIN (C-LYTA) FROM STREP... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1gvm | ||||||
|---|---|---|---|---|---|---|---|
| Title | CHOLINE BINDING DOMAIN OF THE MAJOR AUTOLYSIN (C-LYTA) FROM STREPTOCOCCUS PNEUMONIAE | ||||||
 Components Components | AUTOLYSIN | ||||||
 Keywords Keywords | CHOLINE-BINDING DOMAIN / CELL WALL ATTACHMENT | ||||||
| Function / homology |  Function and homology information Function and homology informationN-acetylmuramoyl-L-alanine amidase / establishment of competence for transformation / N-acetylmuramoyl-L-alanine amidase activity / sporulation resulting in formation of a cellular spore / peptidoglycan catabolic process / cell wall organization / extracellular region Similarity search - Function | ||||||
| Biological species |   STREPTOCOCCUS PNEUMONIAE (bacteria) STREPTOCOCCUS PNEUMONIAE (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.8 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.8 Å | ||||||
 Authors Authors | Fernandez-Tornero, C. / Lopez, R. / Garcia, E. / Gimenez-Gallego, G. / Romero, A. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2002 Title: Two New Crystal Forms of the Choline-Binding Domain of the Major Pneumococcal Autolysin: Insights Into the Dynamics of the Active Dimeric Authors: Fernandez-Tornero, C. / Garcia, E. / Lopez, R. / Gimenez-Gallego, G. / Romero, A. #1: Journal: Nat.Struct.Biol. / Year: 2001Title: A Novel Solenoid Fold in the Cell Wall Anchoring Domain of the Pneumococcal Virulence Factorlyta Authors: Fernandez-Tornero, C. / Lopez, R. / Garcia, E. / Gimenez-Gallego, G. / Romero, A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1gvm.cif.gz | 172.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1gvm.ent.gz | 139.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1gvm.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gv/1gvmftp://data.pdbj.org/pub/pdb/validation_reports/gv/1gvm | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1hcxS S: Starting model for refinement |
|---|---|
| Similar structure data |







-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||||||||
| 2 | 
| ||||||||||||||||||||||||
| 3 | 
| ||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
|
-Components
| #1: Protein | Mass: 15876.638 Da / Num. of mol.: 6 Source method: isolated from a genetically manipulated source Source: (gene. exp.) STREPTOCOCCUS PNEUMONIAE (bacteria) / Plasmid: PCE17 / Production host: References: UniProt: P06653, N-acetylmuramoyl-L-alanine amidase #2: Chemical | ChemComp-CHT / 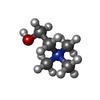  Mass: 104.171 Da / Num. of mol.: 20 / Source method: obtained synthetically / Formula: C5H14NO Mass: 104.171 Da / Num. of mol.: 20 / Source method: obtained synthetically / Formula: C5H14NO#3: Chemical | ChemComp-DDQ / |   Mass: 201.349 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H27NO Mass: 201.349 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H27NO#4: Chemical | ChemComp-TRS / |   Mass: 122.143 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM Mass: 122.143 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 79 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 79 / Source method: isolated from a natural source / Formula: H2OSequence details | ALA D 194, DISORDERED SIDE-CHAIN IN PDB ENTRY. THE FIRST THREE RESIDUES OF EACH CHAIN (GLY185, ...ALA D 194, DISORDERED | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.6 Å3/Da / Density % sol: 52 % | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6.4 Details: 30% PEG 4000, 0.2 M NA-ACETATE, 0.1 M AMMONIUM-ACETATE, PH 6.4, 0.15 M CHOLINE-CL, 0.4 MM DDAO. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 295 K / pH: 6.5 / Method: vapor diffusion, sitting drop | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: MAR scanner 345 mm plate / Detector: IMAGE PLATE / Date: Sep 15, 2001 |
| Radiation | Monochromator: GRAPHITE / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.8→33 Å / Num. obs: 23628 / % possible obs: 98.5 % / Observed criterion σ(I): 8 / Redundancy: 3.2 % / Biso Wilson estimate: 63.8 Å2 / Rmerge(I) obs: 0.09 / Net I/σ(I): 4.5 |
| Reflection shell | Resolution: 2.8→2.95 Å / Redundancy: 3.1 % / Rmerge(I) obs: 0.28 / Mean I/σ(I) obs: 2.4 / % possible all: 98.7 |
| Reflection | *PLUS Lowest resolution: 33 Å / Num. measured all: 74838 / Rmerge(I) obs: 0.09 |
| Reflection shell | *PLUS Highest resolution: 2.8 Å / % possible obs: 98.7 % / Rmerge(I) obs: 0.28 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1HCX Resolution: 2.8→8 Å / Rfactor Rfree error: 0.007 / Data cutoff high absF: 740335.24 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 47.0206 Å2 / ksol: 0.436606 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 39.9 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.8→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.8→2.97 Å / Rfactor Rfree error: 0.021 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.8 Å / % reflection Rfree: 8 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Lowest resolution: 2.95 Å |
