 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1xuf | ||||||
|---|---|---|---|---|---|---|---|
| Title | TRYPSIN-BABIM-ZN+2, PH 8.2 | ||||||
 Components Components | TRYPSIN | ||||||
 Keywords Keywords | SERINE PROTEASE / COMPLEX / TRYPSIN-ZN+2-SMALL MOLECULE LIGAND / DESIGNED SMALL MOLECULE LIGAND WITH NANOMOLAR AFFINITY | ||||||
| Function / homology |  Function and homology information Function and homology informationtrypsin / serpin family protein binding / serine protease inhibitor complex / digestion / endopeptidase activity / serine-type endopeptidase activity / proteolysis / : / metal ion binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 1.9 Å X-RAY DIFFRACTION / Resolution: 1.9 Å | ||||||
 Authors Authors | Katz, B.A. / Clark, J.M. / Finer-Moore, J.S. / Jenkins, T.E. / Johnson, C.R. / Rose, M.J. / Luong, C. / Moore, W.R. / Stroud, R.M. | ||||||
 Citation Citation | Journal: Nature / Year: 1998 Title: Design of potent selective zinc-mediated serine protease inhibitors. Authors: Katz, B.A. / Clark, J.M. / Finer-Moore, J.S. / Jenkins, T.E. / Johnson, C.R. / Ross, M.J. / Luong, C. / Moore, W.R. / Stroud, R.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1xuf.cif.gz | 77.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1xuf.ent.gz | 58 KB | Display | PDB format |
| PDBx/mmJSON format | 1xuf.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/xu/1xufftp://data.pdbj.org/pub/pdb/validation_reports/xu/1xuf | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1c1nC  1c1oC  1c1pC  1c1qC  1c1rC  1c1tC  1c1uC  1c1vC  1c1wC  1c2dC  1c2eC  1c2fC  1c2gC  1c2hC  1c2iC  1c2jC  1c2kC  1c2lC  1c2mC  1xugC  1xuhC  1xuiC  1xujC  1xukC C: citing same article ( |
|---|---|
| Similar structure data |



















-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 23324.287 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Details: TRYPSIN/ZN+2/SMALL MOLECULE LIGAND COMPLEX / Source: (natural) |
|---|---|
| #2: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca |
| #3: Chemical | ChemComp-BAZ / 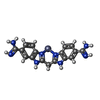  Mass: 397.772 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H16N8Zn Mass: 397.772 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H16N8Zn |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 213 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 213 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.33 Å3/Da / Density % sol: 20 % Description: DATA WITH RSYM > 50 % WERE REJECTED ALONG WITH DATA WHOSE VALUES WERE > 3.5 SIGMA FROM THE MEAN FOR EACH SET OF SYMMETRY EQUIVALENTS |
|---|---|
| Crystal grow | pH: 8.2 Details: SYNTHETIC MOTHER LIQUOR = 75% SATURATED MAGNESIUM SULFATE, 25 % 1.0 M TRIS ADJUSTED TO PH 8.2; SATURATED IN BABIM, 5.0 MM ZN+2. COMPLEX PRODUCED BY SOAKING TRYPSIN-BENZAMIDINE CO-CRYSTAL. |
| Crystal grow | *PLUS Method: unknown |
-Data collection
| Diffraction | Ambient temp details: ROOM |
|---|---|
| Diffraction source | Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS II / Detector: IMAGE PLATE |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Num. obs: 14155 / % possible obs: 57 % / Observed criterion σ(I): 0.5 / Redundancy: 1.6 % / Rmerge(I) obs: 0.085 |
| Reflection shell | Resolution: 2.04→2.13 Å / % possible all: 51 |
| Reflection shell | *PLUS % possible obs: 51 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Highest resolution: 1.9 Å / σ(F): 1
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Highest resolution: 1.9 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 2.04 Å / Lowest resolution: 2.13 Å / Num. reflection Rfree: 2.04 / Num. reflection Rwork: 2.13 |
