- PDB-1uw3: The crystal structure of the globular domain of sheep prion protein -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 1uw3
Title
The crystal structure of the globular domain of sheep prion protein
Components
PRION PROTEIN
Keywords
MEMBRANE PROTEIN / TRANSMISSIBLE SPONGIFORM ENCEPHALOPATHY / CREUTZFELD JACOB DISEASE
Function / homology
Function and homology information
side of membrane / tubulin binding / protein homooligomerization / microtubule binding / copper ion binding / Golgi apparatus / identical protein binding / plasma membrane Similarity search - Function
Prion/Doppel protein, beta-ribbon domain / Major Prion Protein / Prion, copper binding octapeptide repeat / Copper binding octapeptide repeat region / Major prion protein N-terminal domain / Major prion protein bPrPp - N terminal / Prion protein signature 1. / Prion protein signature 2. / Prion protein / Major prion protein ...Prion/Doppel protein, beta-ribbon domain / Major Prion Protein / Prion, copper binding octapeptide repeat / Copper binding octapeptide repeat region / Major prion protein N-terminal domain / Major prion protein bPrPp - N terminal / Prion protein signature 1. / Prion protein signature 2. / Prion protein / Major prion protein / Prion/Doppel protein, beta-ribbon domain / Prion/Doppel beta-ribbon domain superfamily / Prion/Doppel alpha-helical domain / Orthogonal Bundle / Mainly Alpha Similarity search - Domain/homology
Resolution: 2.05→84.51 Å / Cor.coef. Fo:Fc: 0.942 / Cor.coef. Fo:Fc free: 0.829 / SU B: 5.928 / SU ML: 0.167 / Cross valid method: THROUGHOUT / ESU R: 0.279 / ESU R Free: 0.268 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: RESIDUES 125 - 137 ARE POORLY ORDERED, AND PROBABLY TAKE UP MORE THAN ONE CONFORMATION.
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.33
326
4.6 %
RANDOM
Rwork
0.222
-
-
-
obs
0.227
6769
92.4 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK BULK SOLVENT
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj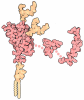



 Assembly
Assembly

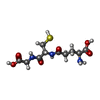
 Mass: 307.323 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H17N3O6S
Mass: 307.323 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H17N3O6S
 Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4
Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 Mass: 18.015 Da / Num. of mol.: 71 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 71 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: PX14.1 / Wavelength: 1.414
/ Beamline: PX14.1 / Wavelength: 1.414  Processing
Processing