Entry Database : PDB / ID : 4aobTitle SAM-I riboswitch containing the T. solenopsae Kt-23 in complex with S- adenosyl methionine SAM-I RIBOSWITCH Keywords / / Function / homology / / / / Biological species THERMOANAEROBACTER TENGCONGENSIS (bacteria)Method / / / Resolution : 2.95 Å Authors Schroeder, K.T. / Daldrop, P. / McPhee, S.A. / Lilley, D.M.J. Journal : RNA / Year : 2012Title : Structure and Folding of a Rare, Natural Kink Turn in RNA with an Aa Pair at the 2B2N Position.Authors : Schroeder, K.T. / Daldrop, P. / Mcphee, S.A. / Lilley, D.M.J. History Deposition Mar 25, 2012 Deposition site / Processing site Revision 1.0 May 9, 2012 Provider / Type Revision 1.1 May 30, 2012 Group Revision 1.2 Dec 20, 2023 Group Data collection / Database references ... Data collection / Database references / Derived calculations / Other / Refinement description Category chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_database_status / pdbx_initial_refinement_model / pdbx_struct_conn_angle / struct_conn / struct_site Item _database_2.pdbx_DOI / _database_2.pdbx_database_accession ... _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_database_status.status_code_sf / _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_asym_id / _pdbx_struct_conn_angle.ptnr1_label_atom_id / _pdbx_struct_conn_angle.ptnr1_label_comp_id / _pdbx_struct_conn_angle.ptnr1_label_seq_id / _pdbx_struct_conn_angle.ptnr1_symmetry / _pdbx_struct_conn_angle.ptnr2_auth_comp_id / _pdbx_struct_conn_angle.ptnr2_auth_seq_id / _pdbx_struct_conn_angle.ptnr2_label_asym_id / _pdbx_struct_conn_angle.ptnr2_label_atom_id / _pdbx_struct_conn_angle.ptnr2_label_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_asym_id / _pdbx_struct_conn_angle.ptnr3_label_atom_id / _pdbx_struct_conn_angle.ptnr3_label_comp_id / _pdbx_struct_conn_angle.ptnr3_label_seq_id / _pdbx_struct_conn_angle.ptnr3_symmetry / _pdbx_struct_conn_angle.value / _struct_conn.pdbx_dist_value / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr1_symmetry / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_conn.ptnr2_label_seq_id / _struct_conn.ptnr2_symmetry / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information THERMOANAEROBACTER TENGCONGENSIS (bacteria)
THERMOANAEROBACTER TENGCONGENSIS (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads








 PDBj
PDBj






























 Assembly
Assembly




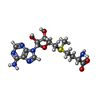

 Mass: 137.327 Da / Num. of mol.: 15 / Source method: obtained synthetically / Formula: Ba
Mass: 137.327 Da / Num. of mol.: 15 / Source method: obtained synthetically / Formula: Ba Mass: 39.098 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: K
Mass: 39.098 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: K Mass: 22.990 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Na
Mass: 22.990 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Na Mass: 398.437 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H22N6O5S
Mass: 398.437 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H22N6O5S Sample preparation
Sample preparation / Beamline: ID14-1 / Wavelength: 0.9334
/ Beamline: ID14-1 / Wavelength: 0.9334  Processing
Processing