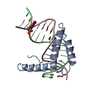
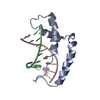
- PDB-1qrv: CRYSTAL STRUCTURE OF THE COMPLEX OF HMG-D AND DNA -
+
データを開く
IDまたはキーワード:
読み込み中...
-
基本情報
登録情報
データベース: PDB / ID: 1qrv
タイトル
CRYSTAL STRUCTURE OF THE COMPLEX OF HMG-D AND DNA
要素
DNA (5'-D(*GP*CP*GP*AP*TP*AP*TP*CP*GP*C)-3')
HIGH MOBILITY GROUP PROTEIN D
キーワード
GENE REGULATION/DNA / PROTEIN-DNA COMPLEX / HMG DOMAIN / NON-SEQUENCE SPECIFIC CHROMOSOMAL PROTEIN HMG-D / GENE REGULATION-DNA COMPLEX
機能・相同性
機能・相同性情報
Mitochondrial transcription initiation / Mitochondrial protein degradation / DNA binding, bending / minor groove of adenine-thymine-rich DNA binding / chromatin organization / regulation of transcription by RNA polymerase II / chromatin / DNA binding / nucleus 類似検索 - 分子機能
: / High mobility group box domain / DNA Binding (I), subunit A / HMG (high mobility group) box / HMG boxes A and B DNA-binding domains profile. / high mobility group / High mobility group box domain / High mobility group box domain superfamily / Orthogonal Bundle / Mainly Alpha 類似検索 - ドメイン・相同性
C: DNA (5'-D(*GP*CP*GP*AP*TP*AP*TP*CP*GP*C)-3') D: DNA (5'-D(*GP*CP*GP*AP*TP*AP*TP*CP*GP*C)-3') A: HIGH MOBILITY GROUP PROTEIN D B: HIGH MOBILITY GROUP PROTEIN D ヘテロ分子
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報
 X線回折 /
X線回折 /  データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード











 PDBj
PDBj

 集合体
集合体


 分子量: 22.990 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Na
分子量: 22.990 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Na 分子量: 18.015 Da / 分子数: 116 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 116 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製
 解析
解析