 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1ppc: GEOMETRY OF BINDING OF THE BENZAMIDINE-AND ARGININE-BASED INHIBIT... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ppc | ||||||
|---|---|---|---|---|---|---|---|
| Title | GEOMETRY OF BINDING OF THE BENZAMIDINE-AND ARGININE-BASED INHIBITORS N-ALPHA-(2-NAPHTHYL-SULPHONYL-GLYCYL)-DL-P-AMIDINOPHENYLALANYL-PIPERIDINE (NAPAP) AND (2R,4R)-4-METHYL-1-[N-ALPHA-(3-METHYL-1,2,3,4-TETRAHYDRO-8-QUINOLINESULPHONYL)-L-ARGINYL]-2-PIPERIDINE CARBOXYLIC ACID (MQPA) TO HUMAN ALPHA-THROMBIN: X-RAY CRYSTALLOGRAPHIC DETERMINATION OF THE NAPAP-TRYPSIN COMPLEX AND MODELING OF NAPAP-THROMBIN AND MQPA-THROMBIN | ||||||
 Components Components | TRYPSIN | ||||||
 Keywords Keywords | HYDROLASE/hydrolase inhibitor / SERINE PROTEINASE / HYDROLASE-hydrolase inhibitor complex | ||||||
| Function / homology |  Function and homology information Function and homology informationtrypsin / serpin family protein binding / serine protease inhibitor complex / digestion / endopeptidase activity / serine-type endopeptidase activity / proteolysis / : / metal ion binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 1.8 Å X-RAY DIFFRACTION / Resolution: 1.8 Å | ||||||
 Authors Authors | Bode, W. / Turk, D. | ||||||
 Citation Citation | Journal: Eur.J.Biochem. / Year: 1990 Title: Geometry of binding of the benzamidine- and arginine-based inhibitors N alpha-(2-naphthyl-sulphonyl-glycyl)-DL-p-amidinophenylalanyl-pipe ridine (NAPAP) and (2R,4R)-4-methyl-1-[N alpha-(3- ...Title: Geometry of binding of the benzamidine- and arginine-based inhibitors N alpha-(2-naphthyl-sulphonyl-glycyl)-DL-p-amidinophenylalanyl-pipe ridine (NAPAP) and (2R,4R)-4-methyl-1-[N alpha-(3-methyl-1,2,3,4-tetrahydr quinolinesulphonyl)-L-arginyl]-2-piperidine carboxylic acid (MQPA) to human alpha-thrombin.X-ray crystallographic determination of the NAPAP-trypsin complex and modeling of NAPAP-thrombin and MQPA-thrombin. Authors: Bode, W. / Turk, D. / Sturzebecher, J. #1: Journal: J.Mol.Biol. / Year: 1992Title: Refined 2.3 Angstroms X-Ray Crystal Structure of Bovine Thrombin Complexes Formed with the Benzamidine and Arginine-Based Thrombin Inhibitors Napap, 4-Tapap and Mqpa. A Starting Point for Improving Antithrombotics Authors: Brandstetter, H. / Turk, D. / Hoeffken, H.W. / Grosse, D. / Stuerzebecher, J. / Martin, P.D. / Edwards, B.F. / Bode, W. #2: Journal: Protein Sci. / Year: 1992Title: The Refined 1.9 Angstroms X-Ray Crystal Structure of D-Phe-Pro-Arg-Chloromethylketone-Inhibited Human Alpha-Thrombin: Structure Analysis, Overall Structure, Electrostatic Properties, Detailed ...Title: The Refined 1.9 Angstroms X-Ray Crystal Structure of D-Phe-Pro-Arg-Chloromethylketone-Inhibited Human Alpha-Thrombin: Structure Analysis, Overall Structure, Electrostatic Properties, Detailed Active-Site Geometry, and Structure-Function Relationships Authors: Bode, W. / Turk, D. / Karshikov, A. #3: Journal: FEBS Lett. / Year: 1991Title: Geometry of Binding of the Nalpha-Tosylated Piperidides of M-Amidino-, P-Amidino-and P-Guanidino Phenylalanine to Thrombin and Trypsin: X-Ray Crystal Structures of Their Trypsin Complexes and ...Title: Geometry of Binding of the Nalpha-Tosylated Piperidides of M-Amidino-, P-Amidino-and P-Guanidino Phenylalanine to Thrombin and Trypsin: X-Ray Crystal Structures of Their Trypsin Complexes and Modeling of Their Thrombin Complexes Authors: Turk, D. / Stuerzebecher, J. / Bode, W. #4: Journal: Embo J. / Year: 1989Title: The Refined 1.9 Angstroms Crystal Structure of Human Alpha-Thrombin: Interaction with D-Phe-Pro-Arg Chloromethylketone and Significance of the Tyr-Pro-Pro-Trp Insertion Segment Authors: Bode, W. / Mayr, I. / Baumann, U. / Huber, R. / Stone, S.R. / Hofsteenge, J. #5: Journal: J.Mol.Biol. / Year: 1989Title: Crystal Structure of Bovine Beta-Trypsin at 1.5 Angstroms Resolution in a Crystal Form with Low Molecular Packing Density. Active Site Geometry, Ion Pairs and Solvent Structure Authors: Bartunik, H.D. / Summers, L.J. / Bartsch, H.H. #6: Journal: J.Mol.Biol. / Year: 1975Title: The Refined Crystal Structure of Bovine Beta-Trypsin at 1.8 Angstroms Resolution. Crystallographic Refinement, Calcium Binding Site, Benzamidine Binding Site and Active Site at Ph 7.0 Authors: Bode, W. / Schwager, P. | ||||||
| History |
|
- Structure visualization
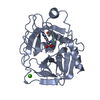
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ppc.cif.gz | 60.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ppc.ent.gz | 42.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1ppc.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/pp/1ppcftp://data.pdbj.org/pub/pdb/validation_reports/pp/1ppc | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|



















-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 23324.287 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
|---|---|
| #2: Chemical | ChemComp-MID / 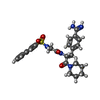  Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 521.631 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H31N5O4S Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 521.631 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H31N5O4SReferences: N(alpha)-(2-naphthylsulfonylglycyl)-4-amidinophenylalanine piperidide |
| #3: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 147 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 147 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
| Nonpolymer details | NAPAP, A SYNTHETIC THROMBIN INHIBITOR, IS NONCOVALENTLY BOUND TO THE ACTIVE SITE. A CALCIUM ION IS ...NAPAP, A SYNTHETIC THROMBIN INHIBITOR, IS NONCOVALEN |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.94 Å3/Da / Density % sol: 58.14 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS Temperature: 20 ℃ / pH: 6 / Method: vapor diffusion | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | *PLUS Highest resolution: 1.8 Å / Num. obs: 21332 / % possible obs: 81 % / Num. measured all: 71775 / Rmerge(I) obs: 0.083 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Rfactor Rwork: 0.181 / Rfactor obs: 0.181 / Highest resolution: 1.8 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Highest resolution: 1.8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.8 Å / Lowest resolution: 8 Å / Num. reflection obs: 21051 / Rfactor obs: 0.181 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 15.4 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: x_angle_d / Dev ideal: 2.54 |
