 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1jrs | ||||||
|---|---|---|---|---|---|---|---|
| Title | HEMIACETAL COMPLEX BETWEEN LEUPEPTIN AND TRYPSIN | ||||||
 Components Components |
| ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / HYDROLASE / SERINE PROTEASE / DIGESTION / PANCREAS / ZYMOGEN / HYDROLASE (SERINE PROTEASE) / HYDROLASE-HYDROLASE INHIBITOR COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationtrypsin / serpin family protein binding / serine protease inhibitor complex / digestion / endopeptidase activity / serine-type endopeptidase activity / proteolysis / : / metal ion binding Similarity search - Function | ||||||
| Biological species |   Actinomycetes Streptomyces roseus MA 839-A1 (bacteria) Actinomycetes Streptomyces roseus MA 839-A1 (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 1.8 Å X-RAY DIFFRACTION / Resolution: 1.8 Å | ||||||
 Authors Authors | Kurinov, I.V. / Harrison, R.W. | ||||||
 Citation Citation | Journal: Protein Sci. / Year: 1996 Title: Two crystal structures of the leupeptin-trypsin complex. Authors: Kurinov, I.V. / Harrison, R.W. #1: Journal: Nat.Struct.Biol. / Year: 1994Title: Prediction of New Serine Proteinase Inhibitors Authors: Kurinov, I.V. / Harrison, R.W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1jrs.cif.gz | 74.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1jrs.ent.gz | 54.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1jrs.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/jr/1jrsftp://data.pdbj.org/pub/pdb/validation_reports/jr/1jrs | HTTPS FTP |
|---|
-Related structure data




















-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 23324.287 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) | ||||
|---|---|---|---|---|---|
| #2: Protein/peptide | 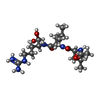  Type: Oligopeptide / Class: Enzyme inhibitor / Mass: 429.578 Da / Num. of mol.: 1 / Source method: obtained synthetically Type: Oligopeptide / Class: Enzyme inhibitor / Mass: 429.578 Da / Num. of mol.: 1 / Source method: obtained syntheticallySource: (synth.) Actinomycetes Streptomyces roseus MA 839-A1 (bacteria)References: NOR: NOR00487, LEUPEPTIN, trypsin | ||||
| #3: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca | ||||
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 170 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 170 / Source method: isolated from a natural source / Formula: H2O | ||||
| Compound details | THE LEUPEPTIN IS COVALENTLY| Has protein modification | Y | Sequence details | THERE IS A DISCREPANCY IN THE NORINE AND PDB NUMBERING, AS NORINE COUNTS ACE AND LEU TOGETHER AS ...THERE IS A DISCREPANC | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.93 Å3/Da / Density % sol: 56 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Method: vapor diffusion, hanging drop Details: ORTHORHOMBIC SPACE GROUP; CRYSTALLIZATION CONDITIONS: 24-26% PEG8000, 0.02M INHIBITOR, 0.2M (NH4)2 SO4, 50MM TRIS-HCL, ROOM TEMPERATURE, HANGING DROP., vapor diffusion - hanging drop Temp details: room temp | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 8 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction source | Wavelength: 1.5418 |
|---|---|
| Detector | Type: RIGAKU / Detector: IMAGE PLATE / Date: Mar 19, 1994 |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Num. obs: 78707 / % possible obs: 88.8 % / Observed criterion σ(I): 1 / Redundancy: 3.24 % / Rmerge(I) obs: 0.044 |
| Reflection | *PLUS Highest resolution: 1.8 Å |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.8→8 Å / σ(F): 2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 22.2 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
