 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1yyy | ||||||
|---|---|---|---|---|---|---|---|
| Title | Trypsin inhibitors with rigid tripeptidyl aldehydes | ||||||
 Components Components | TRYPSIN | ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / SERINE PROTEASE / HYDROLASE-HYDROLASE INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationtrypsin / serpin family protein binding / serine protease inhibitor complex / digestion / endopeptidase activity / serine-type endopeptidase activity / proteolysis / : / metal ion binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.1 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.1 Å | ||||||
 Authors Authors | Krishnan, R. / Zhang, E. / Hakansson, K. / Arni, R.K. / Tulinsky, A. / Lim-Wilby, M.S.L. / Levy, O.E. / Semple, J.E. / Brunck, T.K. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1998 Title: Highly selective mechanism-based thrombin inhibitors: structures of thrombin and trypsin inhibited with rigid peptidyl aldehydes. Authors: Krishnan, R. / Zhang, E. / Hakansson, K. / Arni, R.K. / Tulinsky, A. / Lim-Wilby, M.S. / Levy, O.E. / Semple, J.E. / Brunck, T.K. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1yyy.cif.gz | 61 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1yyy.ent.gz | 42.6 KB | Display | PDB format |
| PDBx/mmJSON format | 1yyy.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/yy/1yyyftp://data.pdbj.org/pub/pdb/validation_reports/yy/1yyy | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1ba8C  1bb0C  1ca8C  1zzzC  1ppcS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 23324.287 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) |
|---|---|
| #2: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca |
| #3: Chemical | ChemComp-0KV / 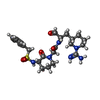  Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 506.618 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H34N6O5S Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 506.618 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H34N6O5SReferences: 2-{(3S)-3-[(benzylsulfonyl)amino]-2-oxopiperidin-1-yl}-N-{(2S)-1-[(3S)-1-carbamimidoylpiperidin-3-yl]-3-oxopropan-2-yl}acetamide |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 157 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 157 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
| Nonpolymer details | THE ACTIVE SITE INHIBITOR IS IN ITS S CHIRAL FORM. THE ACTIVE SITE SER 195 IS CLOSE TO THE CARBONYL ...THE ACTIVE SITE INHIBITOR IS IN ITS S CHIRAL FORM. THE ACTIVE SITE SER 195 IS CLOSE TO THE CARBONYL CARBON (C3) OF THE ALDEHYDE GROUP OF THE INHIBITOR FORMING A TRANSITION |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.99 Å3/Da / Density % sol: 58.85 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7.5 / Details: pH 7.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 7 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 300 K |
|---|---|
| Diffraction source | Wavelength: 1.5418 |
| Detector | Type: RIGAKU / Detector: IMAGE PLATE / Date: Sep 1, 1995 |
| Radiation | Monochromator: NI FILTER / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Highest resolution: 2.1 Å / Num. obs: 13609 / % possible obs: 76 % / Observed criterion σ(I): 2 / Redundancy: 2 % / Rmerge(I) obs: 0.043 / Rsym value: 0.12 / Net I/σ(I): 15.4 |
| Reflection shell | Resolution: 2.1→2.3 Å / Redundancy: 2 % / Rmerge(I) obs: 0.061 / Mean I/σ(I) obs: 2.5 / Rsym value: 0.25 / % possible all: 52 |
| Reflection | *PLUS Redundancy: 2.9 % / Num. measured all: 39107 |
| Reflection shell | *PLUS % possible obs: 52 % / Mean I/σ(I) obs: 10.1 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1PPC Resolution: 2.1→7 Å / Cross valid method: THROUGHOUT / σ(F): 3
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 17.1 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.2 Å / Luzzati d res low obs: 7 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→7 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: PROFFT / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.142 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: p_angle_d / Dev ideal: 0.05 |
