 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1ot6: CRYOTRAPPED CRYSTAL STRUCTURE OF THE E46Q MUTANT OF PHOTOACTIVE Y... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ot6 | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYOTRAPPED CRYSTAL STRUCTURE OF THE E46Q MUTANT OF PHOTOACTIVE YELLOW PROTEIN UNDER CONTINUOUS ILLUMINATION AT 110K | ||||||
 Components Components | Photoactive yellow protein | ||||||
 Keywords Keywords | SIGNALING PROTEIN / PYP / cryotrapping | ||||||
| Function / homology |  Function and homology information Function and homology informationphotoreceptor activity / phototransduction / regulation of DNA-templated transcription / identical protein binding Similarity search - Function | ||||||
| Biological species |  Halorhodospira halophila (bacteria) Halorhodospira halophila (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 0.95 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 0.95 Å | ||||||
 Authors Authors | Anderson, S. / Crosson, S. / Moffat, K. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2004 Title: Short hydrogen bonds in photoactive yellow protein. Authors: Anderson, S. / Crosson, S. / Moffat, K. | ||||||
| History |
| ||||||
| Remark 300 | BIOMOLECULE Model 1 is associated with the photoactivated state and Model 2 is associated with the ...BIOMOLECULE Model 1 is associated with the photoactivated state and Model 2 is associated with the ground state of the protein. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ot6.cif.gz | 120.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ot6.ent.gz | 99.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1ot6.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ot/1ot6ftp://data.pdbj.org/pub/pdb/validation_reports/ot/1ot6 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1ot9C  1otaC  1otbC  1otdC  1oteC  1otiC C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Number of models | 2 |
-Components
| #1: Protein | Mass: 13887.591 Da / Num. of mol.: 1 / Mutation: E46Q Source method: isolated from a genetically manipulated source Source: (gene. exp.) Halorhodospira halophila (bacteria) / Gene: PYP / Production host: |
|---|---|
| #2: Chemical | ChemComp-HC4 / 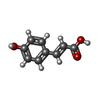  Mass: 164.158 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H8O3 Mass: 164.158 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H8O3 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 185 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 185 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.84 Å3/Da / Density % sol: 33.28 % Description: Author collected the first dataset in the dark, then a second dataset was collected on the same crystal after inducing a photostationary state with continuous illumination. The structure ...Description: Author collected the first dataset in the dark, then a second dataset was collected on the same crystal after inducing a photostationary state with continuous illumination. The structure was refined by the application of difference refinement to both datasets. |
|---|---|
| Crystal grow | Method: vapor diffusion, hanging drop / pH: 7 Details: ammonium sulphate, sodium phosphate, pH 7.0, VAPOR DIFFUSION, HANGING DROP, temperature 110K |
-Data collection
| Diffraction |
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 14-BM-C / Wavelength: 1 Å / Beamline: 14-BM-C / Wavelength: 1 Å | |||||||||||||||
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Feb 26, 2001 | |||||||||||||||
| Radiation |
| |||||||||||||||
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 | |||||||||||||||
| Reflection | Resolution: 0.95→100 Å / Num. all: 63595 / Num. obs: 63595 / % possible obs: 93.5 % / Observed criterion σ(F): 2 / Observed criterion σ(I): 2 / Redundancy: 13.5 % / Rmerge(I) obs: 0.046 / Net I/σ(I): 26.5 | |||||||||||||||
| Reflection shell | Resolution: 0.95→0.98 Å / Rmerge(I) obs: 0.254 / Mean I/σ(I) obs: 26.5 / % possible all: 78.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 0.95→20 Å / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 0.95→20 Å
| ||||||||||||||||||||
| Refine LS restraints |
|
