 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3pyp: PHOTOACTIVE YELLOW PROTEIN, CRYOTRAPPED EARLY LIGHT CYCLE INTERMEDIATE -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3pyp | ||||||
|---|---|---|---|---|---|---|---|
| Title | PHOTOACTIVE YELLOW PROTEIN, CRYOTRAPPED EARLY LIGHT CYCLE INTERMEDIATE | ||||||
 Components Components | PHOTOACTIVE YELLOW PROTEIN | ||||||
 Keywords Keywords | PHOTORECEPTOR / LIGHT SENSOR FOR NEGATIVE PHOTOTAXIS | ||||||
| Function / homology |  Function and homology information Function and homology informationphotoreceptor activity / phototransduction / regulation of DNA-templated transcription / identical protein binding Similarity search - Function | ||||||
| Biological species |  Halorhodospira halophila (bacteria) Halorhodospira halophila (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / SAME CRYSTAL LATTICE MOLECULAR REPLACEMENT / Resolution: 0.85 Å X-RAY DIFFRACTION / SYNCHROTRON / SAME CRYSTAL LATTICE MOLECULAR REPLACEMENT / Resolution: 0.85 Å | ||||||
 Authors Authors | Genick, U.K. / Soltis, S.M. / Kuhn, P. / Canestrelli, I.L. / Getzoff, E.D. | ||||||
 Citation Citation | Journal: Nature / Year: 1998 Title: Structure at 0.85 A resolution of an early protein photocycle intermediate. Authors: Genick, U.K. / Soltis, S.M. / Kuhn, P. / Canestrelli, I.L. / Getzoff, E.D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3pyp.cif.gz | 105.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3pyp.ent.gz | 82.9 KB | Display | PDB format |
| PDBx/mmJSON format | 3pyp.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/py/3pypftp://data.pdbj.org/pub/pdb/validation_reports/py/3pyp | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2phyS S: Starting model for refinement |
|---|---|
| Similar structure data |






-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
| ||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 13888.575 Da / Num. of mol.: 1 / Source method: isolated from a natural source Details: THIOESTER LINKAGE BETWEEN THE SULFUR OF CYS 69 AND CARBOXY GROUP OF THE 4-HYDROXY CINNAMIC ACID CHROMOPHORE Source: (natural) Halorhodospira halophila (bacteria) / Strain: BN9626 / References: UniProt: P16113 |
|---|---|
| #2: Chemical | ChemComp-HC4 / 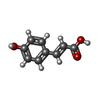  Mass: 164.158 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H8O3 Mass: 164.158 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H8O3 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 137 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 137 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.8 Å3/Da / Density % sol: 32 % / Description: CRYSTALS IDENTICAL TO 2PHY | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 4.8 / Details: pH 4.8 | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 7 / Method: vapor diffusion, hanging drop / Details: used as seeds | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 149 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL9-1 / Wavelength: 0.77 / Beamline: BL9-1 / Wavelength: 0.77 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Sep 6, 1997 / Details: MIRRORS |
| Radiation | Monochromator: SI(111) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.77 Å / Relative weight: 1 |
| Reflection | Highest resolution: 0.85 Å / Num. obs: 84934 / % possible obs: 97.7 % / Observed criterion σ(I): 0 / Redundancy: 6.6 % / Rmerge(I) obs: 0.042 / Rsym value: 0.042 / Net I/σ(I): 10.9 |
| Reflection shell | Resolution: 0.85→0.86 Å / Rmerge(I) obs: 0.52 / Mean I/σ(I) obs: 1.7 / Rsym value: 0.52 / % possible all: 87.1 |
| Reflection | *PLUS Num. measured all: 561693 |
| Reflection shell | *PLUS % possible obs: 87.1 % |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAME CRYSTAL LATTICE MOLECULAR REPLACEMENT Starting model: 2PHY Resolution: 0.85→50 Å / Num. parameters: 11379 / Num. restraintsaints: 15920 / Cross valid method: FREE R-VALUE / σ(F): 0 / Stereochemistry target values: ENGH AND HUBER Details: NUMBER OF PROTEIN AND SOLVENT ATOMS CONTAINS DUAL CONFORMERS
| |||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: MOEWS & KRETSINGER | |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 18 / Occupancy sum hydrogen: 943 / Occupancy sum non hydrogen: 1133 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 0.85→50 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
