 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1nc2: Crystal Structure of Monoclonal Antibody 2D12.5 Fab Complexed wit... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1nc2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of Monoclonal Antibody 2D12.5 Fab Complexed with Y-DOTA | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | IMMUNE SYSTEM / ANTIBODY-DOTA COMPLEX / RARE EARTH / DOTA / METAL CHELATE / YTTRIUM / GAMMA TURN / N-LINKED GLYCOSYLATION | |||||||||
| Function / homology | Immunoglobulins / Immunoglobulin-like / Sandwich / Mainly Beta / Chem-DOE / YTTRIUM (III) ION Function and homology information Function and homology information | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å | |||||||||
 Authors Authors | Corneillie, T.M. / Fisher, A.J. / Meares, C.F. | |||||||||
 Citation Citation | Journal: J.Am.Chem.Soc. / Year: 2003 Title: Crystal structures of two complexes of the rare-earth-DOTA-binding antibody 2D12.5: ligand generality from a chiral system. Authors: Corneillie, T.M. / Fisher, A.J. / Meares, C.F. | |||||||||
| History |
| |||||||||
| Remark 999 | SEQUENCE AUTHORS INFORMED THAT THE SEQUENCE FOR THIS PROTEIN IS NOT YET AVAILBLE IN ANY REFERENCE ...SEQUENCE AUTHORS INFORMED THAT THE SEQUENCE FOR THIS PROTEIN IS NOT YET AVAILBLE IN ANY REFERENCE SEQUENCE DATABASE. Actual amino acid sequence positions are used for the atomic coordinates. Kabat numbering is used in the cited journal article. An alignment of Kabat position vs. actual amino acid sequence position is available in the supporting information that accompanies the article. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1nc2.cif.gz | 184.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1nc2.ent.gz | 145.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1nc2.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/nc/1nc2ftp://data.pdbj.org/pub/pdb/validation_reports/nc/1nc2 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1nc4C  1gigS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
-Antibody , 3 types, 4 molecules ACBD
| #1: Antibody | Mass: 23150.602 Da / Num. of mol.: 2 / Fragment: Fab / Source method: isolated from a natural source / Source: (natural) Cell line: MYELOMA LINE PX63 AG8 FUSED WITH ANTIBODY EXPRESSING SPLEEN CELLS Strain: BALB/c #2: Antibody | | Mass: 23762.586 Da / Num. of mol.: 1 / Fragment: Fab / Source method: isolated from a natural source / Source: (natural) Cell line: MYELOMA LINE PX63 AG8 FUSED WITH ANTIBODY EXPRESSING SPLEEN CELLS Strain: BALB/c #3: Antibody | | Mass: 23779.615 Da / Num. of mol.: 1 / Fragment: Fab / Source method: isolated from a natural source / Source: (natural) Cell line: MYELOMA LINE PX63 AG8 FUSED WITH ANTIBODY EXPRESSING SPLEEN CELLS Strain: BALB/c |
|---|
-Sugars , 1 types, 1 molecules 
| #7: Sugar | ChemComp-NAG /  Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 1 Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 1Source method: isolated from a genetically manipulated source Formula: C8H15NO6 |
|---|
-Non-polymers , 4 types, 368 molecules 

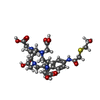

| #4: Chemical |  Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl#5: Chemical |  Mass: 88.906 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Y Mass: 88.906 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Y#6: Chemical |  Mass: 627.707 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C27H41N5O10S Mass: 627.707 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C27H41N5O10S#8: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 362 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.25 Å3/Da / Density % sol: 44.96 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: PEG 8000, HEPES, sodium chloride, pH 7.5, VAPOR DIFFUSION, HANGING DROP, temperature 295.0K | ||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 290 K / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL9-2 / Wavelength: 0.98 Å / Beamline: BL9-2 / Wavelength: 0.98 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: May 1, 2001 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.98 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→30 Å / Num. all: 50304 / Num. obs: 50304 / % possible obs: 95.9 % / Observed criterion σ(F): -1.7 / Observed criterion σ(I): -3 / Redundancy: 2.53 % / Biso Wilson estimate: 17 Å2 / Rmerge(I) obs: 0.088 / Net I/σ(I): 10.7 |
| Reflection shell | Resolution: 2.1→2.18 Å / Redundancy: 2.38 % / Rmerge(I) obs: 0.299 / Mean I/σ(I) obs: 2.9 / Num. unique all: 4899 / % possible all: 95 |
| Reflection | *PLUS Lowest resolution: 30 Å / Num. obs: 50276 / Num. measured all: 127387 |
| Reflection shell | *PLUS % possible obs: 95 % / Num. unique obs: 4899 / Num. measured obs: 11654 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1GIG Resolution: 2.1→29.2 Å / Rfactor Rfree error: 0.005 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber
| |||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 20.7875 Å2 / ksol: 0.338511 e/Å3 | |||||||||||||||||||||||||
| Displacement parameters | Biso mean: 29.7 Å2
| |||||||||||||||||||||||||
| Refine analyze |
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→29.2 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.1→2.23 Å / Rfactor Rfree error: 0.014 / Total num. of bins used: 6
| |||||||||||||||||||||||||
| Xplor file |
| |||||||||||||||||||||||||
| Refinement | *PLUS % reflection Rfree: 5 % | |||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
