 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1m7o: Plasmodium Falciparum Triosephosphate isomerase (PfTIM) compled t... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1m7o | ||||||
|---|---|---|---|---|---|---|---|
| Title | Plasmodium Falciparum Triosephosphate isomerase (PfTIM) compled to substrate analog 3-phosphoglycerate (3PG) | ||||||
 Components Components | Triosephosphate Isomerase | ||||||
 Keywords Keywords | ISOMERASE / TIM barrels / beta-alpha barrels / enzyme-inhibitor complex | ||||||
| Function / homology |  Function and homology information Function and homology informationtriose-phosphate isomerase / triose-phosphate isomerase activity / glyceraldehyde-3-phosphate biosynthetic process / glycerol catabolic process / gluconeogenesis / glycolytic process / identical protein binding / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.4 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.4 Å | ||||||
 Authors Authors | Parthasarathy, S. / Balaram, H. / Balaram, P. / Murthy, M.R.N. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2002 Title: Structures of Plasmodium falciparum triosephosphate isomerase complexed to substrate analogues: observation of the catalytic loop in the open conformation in the ligand-bound state. Authors: Parthasarathy, S. / Balaram, H. / Balaram, P. / Murthy, M.R. #1: Journal: Structure / Year: 1997Title: Triosephosphate Isomerase From Plasmodium Falciparum: Crystal Structure Proveides Insights into Antimalarial Drug Design. Authors: Velankar, S.S. / Ray, S.S. / Gokle, R.S. / Suma, S. / Balaram, H. / Balaram, P. / Murthy, M.R.N. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1m7o.cif.gz | 110.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1m7o.ent.gz | 86.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1m7o.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/m7/1m7oftp://data.pdbj.org/pub/pdb/validation_reports/m7/1m7o | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1m7pC  1ydvS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |






















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 27997.738 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Gene: TPI / Plasmid: ptrc 99A vector, called pARC / Production host:  #2: Chemical | 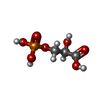  Mass: 186.057 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H7O7P Mass: 186.057 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H7O7P#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 130 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 130 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.26 Å3/Da / Density % sol: 43.5 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 4.5 Details: 12% to 20% PEG 1450 in 100mm Sodium acetate, pH 4.5, VAPOR DIFFUSION, HANGING DROP at 295K | ||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 7.5 | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 295 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 Å |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Jun 9, 1999 / Details: Mirrors |
| Radiation | Monochromator: Mirrors / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.4→20 Å / Num. obs: 19146 / % possible obs: 97.3 % / Observed criterion σ(I): 0 / Redundancy: 3.8 % / Biso Wilson estimate: 18.3 Å2 / Rsym value: 0.116 / Net I/σ(I): 10.5 |
| Reflection shell | Resolution: 2.4→2.49 Å / Redundancy: 3.7 % / Mean I/σ(I) obs: 3.7 / Num. unique all: 1868 / Rsym value: 0.399 / % possible all: 95.3 |
| Reflection | *PLUS Highest resolution: 2.4 Å / Num. measured all: 73117 / Rmerge(I) obs: 0.116 |
| Reflection shell | *PLUS % possible obs: 95.3 % / Rmerge(I) obs: 0.399 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: Unbound PfTIM; PDB CODE 1YDV Resolution: 2.4→20 Å / Rfactor Rfree error: 0.005 / Data cutoff high absF: 165654.6 / Data cutoff high rms absF: 165654.6 / Data cutoff low absF: 0.1 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0.1 / σ(I): 165654.6 / Stereochemistry target values: Engh & Huber Details: Maximum Likelihood in Amplitute (MLF) is employed. Anisotropic B-value scaling, Bulk solvent correction and 2-fold NCS restraint were used throughout the refinement.
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Bsol: 31.1851 Å2 / ksol: 0.316133 e/Å3 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 23.7 Å2
| ||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→20 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.4→2.55 Å / Rfactor Rfree error: 0.017 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.4 Å / Rfactor Rfree: 0.226 / Rfactor Rwork: 0.182 | ||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.29 / Rfactor Rwork: 0.234 |
