 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1lzo: Plasmodium Falciparum Triosephosphate Isomerase-Phosphoglycolate ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1lzo | ||||||
|---|---|---|---|---|---|---|---|
| Title | Plasmodium Falciparum Triosephosphate Isomerase-Phosphoglycolate Complex | ||||||
 Components Components | Triosephosphate Isomerase | ||||||
 Keywords Keywords | ISOMERASE / Triosephosphate Isomerase / Phosphoglycolate / Loop Dynamics / Loop Open Conformation | ||||||
| Function / homology |  Function and homology information Function and homology informationtriose-phosphate isomerase / triose-phosphate isomerase activity / glyceraldehyde-3-phosphate biosynthetic process / glycerol catabolic process / gluconeogenesis / glycolytic process / identical protein binding / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.8 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.8 Å | ||||||
 Authors Authors | Parthasarathy, S. / Balaram, H. / Balaram, P. / Murthy, M.R. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2002 Title: Structure of the Plasmodium falciparum triosephosphate isomerase-phosphoglycolate complex in two crystal forms: characterization of catalytic loop open and closed conformations in the ligand-bound state Authors: Parthasarathy, S. / Ravindra, G. / Balaram, H. / Balaram, P. / Murthy, M.R. #1: Journal: Structure / Year: 1997Title: Triosephosphate isomerase from Plasmodium falciparum: Crystal structure provides insights into antimalarial drug design Authors: Velankar, S.S. / Ray, S.S. / Gokhle, R.S. / Suma, S. / Balaram, H. / Balaram, P. / Murthy, M.R.N. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1lzo.cif.gz | 183.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1lzo.ent.gz | 149.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1lzo.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/lz/1lzoftp://data.pdbj.org/pub/pdb/validation_reports/lz/1lzo | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1lyxC  1ydvS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||
| 2 | 
| ||||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 27997.738 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Gene: TPI / Plasmid: ptrc 99A vector, called pARC / Production host:  #2: Chemical | 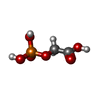  Mass: 156.031 Da / Num. of mol.: 2 Mass: 156.031 Da / Num. of mol.: 2Source method: isolated from a genetically manipulated source Formula: C2H5O6P #3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 150 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 150 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.209 Å3/Da / Density % sol: 42.18 % | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: 12% to 25% PEG 6000 or PEG 4000 or PEG 3350 or PEG 1450 in the presence of EDTA, DTT and Sodium azide in 100mM MES. pH 6.5, VAPOR DIFFUSION, HANGING DROP at 295K | |||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 Å |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Feb 10, 2000 / Details: Mirrors |
| Radiation | Monochromator: Mirrors / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.8→20 Å / Num. all: 24992 / Num. obs: 24992 / % possible obs: 99.5 % / Redundancy: 3.6 % / Biso Wilson estimate: 38.1 Å2 / Rsym value: 0.12 / Net I/σ(I): 10.5 |
| Reflection shell | Resolution: 2.8→2.9 Å / Redundancy: 3 % / Mean I/σ(I) obs: 4.5 / Num. unique all: 2425 / Rsym value: 0.314 / % possible all: 98.5 |
| Reflection | *PLUS Highest resolution: 2.8 Å / Num. obs: 23550 / Num. measured all: 84748 / Rmerge(I) obs: 0.115 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: Dimer of unbound PfTIM, PDB code 1YDV Resolution: 2.8→20 Å / Rfactor Rfree error: 0.006 / Data cutoff high absF: 158207.41 / Data cutoff low absF: 0 Isotropic thermal model: Grouped B factor for main chain and side chain atom of individual residues Cross valid method: THROUGHOUT / σ(F): 0.1 / Stereochemistry target values: Engh & Huber Details: RESOLUTION DEPENDENT WEIGHTING SCHEME, BULK SOLVENT MODEL USED
| |||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 15.4 Å2 / ksol: 0.293485 e/Å3 | |||||||||||||||||||||||||
| Displacement parameters | Biso mean: 14.32 Å2
| |||||||||||||||||||||||||
| Refine analyze |
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.8→20 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| Refine LS restraints NCS | NCS model details: FOUR FOLD NCS RESTRAINTS BETWEEN MONOMERS APPLIED Rms dev Biso : 2 Å2 / Weight Biso : 250 | |||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.8→2.9 Å / Total num. of bins used: 10
| |||||||||||||||||||||||||
| Xplor file |
| |||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.8 Å / Rfactor Rfree: 0.311 / Rfactor Rwork: 0.232 | |||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
