 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1hvc: CRYSTAL STRUCTURE OF A TETHERED DIMER OF HIV-1 PROTEASE COMPLEXED... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1hvc | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF A TETHERED DIMER OF HIV-1 PROTEASE COMPLEXED WITH AN INHIBITOR | ||||||
 Components Components | HIV-1 PROTEASE | ||||||
 Keywords Keywords | HYDROLASE(ACID PROTEASE) | ||||||
| Function / homology |  Function and homology information Function and homology informationviral genome integration into host DNA / establishment of integrated proviral latency / host multivesicular body / RNA-directed DNA polymerase activity / endonuclease activity / aspartic-type endopeptidase activity / virion membrane / proteolysis / DNA binding / zinc ion binding Similarity search - Function | ||||||
| Biological species |   Human immunodeficiency virus 1 Human immunodeficiency virus 1 | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 1.8 Å X-RAY DIFFRACTION / Resolution: 1.8 Å | ||||||
 Authors Authors | Bhat, T.N. / Baldwin, E.T. / Erickson, J.W. | ||||||
 Citation Citation | Journal: Nat.Struct.Biol. / Year: 1994 Title: Crystal structure of a tethered dimer of HIV-1 proteinase complexed with an inhibitor. Authors: Bhat, T.N. / Baldwin, E.T. / Liu, B. / Cheng, Y.S. / Erickson, J.W. #1: Journal: J.Am.Chem.Soc. / Year: 1994Title: Influence of Stereochemistry on Activity and Binding Modes for C2 Symmetry-Based Diol Inhibitors of HIV-1 Protease Authors: Hosur, M.V. / Bhat, T.N. / Kempf, D.J. / Baldwin, E.T. / Liu, B. / Gulnik, S. / Wideburg, N.E. / Norbeck, D.W. / Appelt, K. / Erickson, J.W. #2: Journal: Annu.Rev.Biochem. / Year: 1993Title: Structure-Based Inhibitors of HIV-1 Protease Authors: Wlodawer, A. / Erickson, J.W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1hvc.cif.gz | 89.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1hvc.ent.gz | 68.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1hvc.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hv/1hvcftp://data.pdbj.org/pub/pdb/validation_reports/hv/1hvc | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|






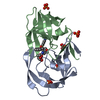



-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Atom site foot note | 1: ILE 15B - GLY 16B OMEGA = 216.69 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 2: SER 203 - GLY 204 OMEGA = 0.34 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 3: ILE 15A - GLY 16A OMEGA = 210.89 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 4: GLY 16A - GLY 17A OMEGA = 348.40 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION |
-Components
| #1: Protein | Mass: 21961.850 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Human immunodeficiency virus 1 / Genus: Lentivirus / Gene: SYNTHETIC GENE / Production host:  |
|---|---|
| #2: Chemical | ChemComp-A79 / 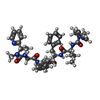  Mass: 794.981 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C44H58N8O6 Mass: 794.981 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C44H58N8O6 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 68 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 68 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.2 Å3/Da / Density % sol: 44.21 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop / Details: using macroseeding | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | *PLUS Highest resolution: 1.8 Å / Lowest resolution: 10 Å / Num. obs: 16421 / % possible obs: 88 % / Num. measured all: 39662 / Rmerge(I) obs: 0.064 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.8→10 Å /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: x_angle_d / Dev ideal: 3 |
