登録情報 データベース : PDB / ID : 1gsjタイトル Selenomethionine substituted N-acetyl-L-glutamate kinase from Escherichia coli complexed with its substrate n-acetyl-L-glutamate and its substrate analog AMPPNP ACETYLGLUTAMATE KINASE キーワード / / / / / / / 機能・相同性 分子機能 ドメイン・相同性 構成要素
/ / / / / / / / / / / / / / / / / / 生物種 ESCHERICHIA COLI BL21 (大腸菌)手法 / / / 解像度 : 1.85 Å データ登録者 Ramon-Maiques, S. / Marina, A. / Gil-Ortiz, F. / Fita, I. / Rubio, V. ジャーナル : Structure / 年 : 2002タイトル : Structure of Acetylglutamate Kinase, a Key Enzyme for Arginine Biosynthesis and a Prototype for the Amino Acid Kinase Enzyme Family, During Catalysis著者 : Ramon-Maiques, S. / Marina, A. / Gil-Ortiz, F. / Fita, I. / Rubio, V. 履歴 登録 2002年1月7日 登録サイト / 処理サイト 改定 1.0 2002年5月16日 Provider / タイプ 改定 1.1 2011年5月8日 Group 改定 1.2 2011年7月13日 Group 改定 1.3 2018年10月24日 Group / Source and taxonomy / カテゴリ / entity_src_genItem _diffrn_source.pdbx_synchrotron_site / _entity_src_gen.gene_src_strain ... _diffrn_source.pdbx_synchrotron_site / _entity_src_gen.gene_src_strain / _entity_src_gen.pdbx_gene_src_ncbi_taxonomy_id / _entity_src_gen.pdbx_gene_src_scientific_name 改定 1.4 2019年7月10日 Group / Derived calculations / カテゴリ / struct_connItem / _struct_conn.pdbx_leaving_atom_flag改定 1.5 2019年7月24日 Group / カテゴリ / Item 改定 1.6 2024年11月13日 Group Data collection / Database references ... Data collection / Database references / Derived calculations / Other / Structure summary カテゴリ chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_database_status / pdbx_entry_details / pdbx_modification_feature / pdbx_struct_conn_angle / struct_conn Item _database_2.pdbx_DOI / _database_2.pdbx_database_accession ... _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_database_status.status_code_sf / _pdbx_entry_details.has_protein_modification / _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_asym_id / _pdbx_struct_conn_angle.ptnr1_label_atom_id / _pdbx_struct_conn_angle.ptnr1_label_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_asym_id / _pdbx_struct_conn_angle.ptnr3_label_atom_id / _pdbx_struct_conn_angle.ptnr3_label_comp_id / _pdbx_struct_conn_angle.value / _struct_conn.pdbx_dist_value / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id
すべて表示 表示を減らす
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報
 X線回折 /
X線回折 /  データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード










 PDBj
PDBj











 集合体
集合体

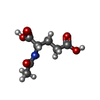
 分子量: 189.166 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C7H11NO5
分子量: 189.166 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C7H11NO5
 分子量: 506.196 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C10H17N6O12P3
分子量: 506.196 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C10H17N6O12P3
 分子量: 24.305 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Mg
分子量: 24.305 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Mg 分子量: 18.015 Da / 分子数: 198 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 198 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: X31 / 波長: 0.94, 0.9765, 0.9770
/ ビームライン: X31 / 波長: 0.94, 0.9765, 0.9770 解析
解析