 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1gfz: FLAVOPIRIDOL INHIBITS GLYCOGEN PHOSPHORYLASE BY BINDING AT THE IN... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1gfz | ||||||
|---|---|---|---|---|---|---|---|
| Title | FLAVOPIRIDOL INHIBITS GLYCOGEN PHOSPHORYLASE BY BINDING AT THE INHIBITOR SITE | ||||||
 Components Components | GLYCOGEN PHOSPHORYLASE | ||||||
 Keywords Keywords | TRANSFERASE / GLYCOGEN PHOSPHORYLASE / GLYCOGEN METABOLISM / DIABETES / FLAVOPIRIDOL / INHIBITOR SITE | ||||||
| Function / homology |  Function and homology information Function and homology informationglycogen phosphorylase / glycogen phosphorylase activity / glycogen catabolic process / skeletal muscle myofibril / pyridoxal phosphate binding / nucleotide binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / OTHER / Resolution: 2.3 Å X-RAY DIFFRACTION / OTHER / Resolution: 2.3 Å | ||||||
 Authors Authors | Oikonomakos, N.G. / Zographos, S.E. / Skamnaki, V.T. / Tsitsanou, K.E. / Johnson, L.N. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2000 Title: Flavopiridol inhibits glycogen phosphorylase by binding at the inhibitor site. Authors: Oikonomakos, N.G. / Schnier, J.B. / Zographos, S.E. / Skamnaki, V.T. / Tsitsanou, K.E. / Johnson, L.N. #1: Journal: Structure / Year: 2000Title: A New Allosteric Site in Glycogen Phosphorylase b as a Target for Drug Interactions Authors: Oikonomakos, N.G. / Skamnaki, V.T. / Tsitsanou, K.E. / Gavalas, N.G. / Johnson, L.N. #2: Journal: Protein Sci. / Year: 1999Title: Allosteric Inhibition of Glycogen Phosphorylase a by the Potential Antidiabetic Drug 3-Isopropyl 4-(2-Chlorophenyl)-1,4-Dihydro-1-Ethyl-2-Methyl-Pyridine-3,5,6-Tricarboxylate Authors: Oikonomakos, N.G. / Tsitsanou, K.E. / Zographos, S.E. / Skamnaki, V.T. / Goldmann, S. / Bischoff, H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1gfz.cif.gz | 180.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1gfz.ent.gz | 142.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1gfz.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gf/1gfzftp://data.pdbj.org/pub/pdb/validation_reports/gf/1gfz | HTTPS FTP |
|---|
-Related structure data






















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 97291.203 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) |
|---|---|
| #2: Chemical | ChemComp-PLP /   Mass: 247.142 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H10NO6P Mass: 247.142 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H10NO6P |
| #3: Chemical | ChemComp-IMP / 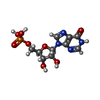  Mass: 348.206 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H13N4O8P Mass: 348.206 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H13N4O8P |
| #4: Chemical | ChemComp-CFF / 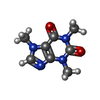  Mass: 194.191 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H10N4O2 / Comment: medication*YM Mass: 194.191 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H10N4O2 / Comment: medication*YM |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 203 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 203 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.46 Å3/Da / Density % sol: 48 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Method: vapor diffusion, hanging drop / pH: 6.7 Details: A single native T-state GPb crystal was soaked in a solution containing 5 mM caffeine, 10 mM Bes, 0.1 mM EDTA buffer, pH 6.7 for 80 min, pH 6.70, VAPOR DIFFUSION, HANGING DROP | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: unknown | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH3R / Wavelength: 1.5418 / Wavelength: 1.5418 Å |
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE / Date: Jun 27, 2000 |
| Radiation | Monochromator: NI FILTER / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.3→28.75 Å / Num. obs: 43063 / % possible obs: 98.3 % / Redundancy: 4.8 % / Rmerge(I) obs: 0.1 / Net I/σ(I): 10.8 |
| Reflection shell | Resolution: 2.3→2.34 Å / Rmerge(I) obs: 0.412 / Mean I/σ(I) obs: 2.8 / % possible all: 94 |
| Reflection | *PLUS Num. measured all: 310740 / Rmerge(I) obs: 0.1 |
| Reflection shell | *PLUS % possible obs: 94 % |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: OTHER / Resolution: 2.3→28.75 Å / Cross valid method: R FREE / σ(F): 0 Details: the final model contains residues 13-256, 261-316, and 324-837. Residues where B-factor values of main chain atoms exceed 60^2 include 13-22, 209-211, 250-256, 313-316, 324-325, 550-557, and ...Details: the final model contains residues 13-256, 261-316, and 324-837. Residues where B-factor values of main chain atoms exceed 60^2 include 13-22, 209-211, 250-256, 313-316, 324-325, 550-557, and 831-837. Thes same regions are also poorly ordered in the native enzyme structure.
| |||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→28.75 Å
| |||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.8 / Classification: refinement | |||||||||||||||||||||||||||
| Refinement | *PLUS | |||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 33.8 Å2 | |||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| |||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.367 / Rfactor Rwork: 0.328 |
