 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 1d4l | ||||||
|---|---|---|---|---|---|---|---|
| タイトル | HIV-1 PROTEASE COMPLEXED WITH A MACROCYCLIC PEPTIDOMIMETIC INHIBITOR | ||||||
 要素 要素 | HIV-1 PROTEASE | ||||||
 キーワード キーワード | HYDROLASE / HIV / PROTEASE / INHIBITOR / ANTIVIRAL | ||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報HIV-1 retropepsin / symbiont-mediated activation of host apoptosis / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / host multivesicular body / DNA integration / viral genome integration into host DNA / RNA-directed DNA polymerase / establishment of integrated proviral latency ...HIV-1 retropepsin / symbiont-mediated activation of host apoptosis / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / host multivesicular body / DNA integration / viral genome integration into host DNA / RNA-directed DNA polymerase / establishment of integrated proviral latency / viral penetration into host nucleus / RNA stem-loop binding / RNA-directed DNA polymerase activity / RNA-DNA hybrid ribonuclease activity / 転移酵素; リンを含む基を移すもの; 核酸を移すもの / host cell / viral nucleocapsid / DNA recombination / DNA-directed DNA polymerase / aspartic-type endopeptidase activity / 加水分解酵素; エステル加水分解酵素 / DNA-directed DNA polymerase activity / symbiont-mediated suppression of host gene expression / viral translational frameshifting / lipid binding / symbiont entry into host cell / host cell nucleus / host cell plasma membrane / virion membrane / structural molecule activity / proteolysis / DNA binding / zinc ion binding / membrane 類似検索 - 分子機能 | ||||||
| 手法 |  X線回折 / 解像度: 1.75 Å X線回折 / 解像度: 1.75 Å | ||||||
 データ登録者 データ登録者 | Tyndall, J.D. / Reid, R.C. / Tyssen, D.P. / Jardine, D.K. / Todd, B. / Passmore, M. / March, D.R. / Pattenden, L.K. / Alewood, D. / Hu, S.H. ...Tyndall, J.D. / Reid, R.C. / Tyssen, D.P. / Jardine, D.K. / Todd, B. / Passmore, M. / March, D.R. / Pattenden, L.K. / Alewood, D. / Hu, S.H. / Alewood, P.F. / Birch, C.J. / Martin, J.L. / Fairlie, D.P. | ||||||
 引用 引用 | ジャーナル: J.Med.Chem. / 年: 2000 タイトル: Synthesis, stability, antiviral activity, and protease-bound structures of substrate-mimicking constrained macrocyclic inhibitors of HIV-1 protease. 著者: Tyndall, J.D. / Reid, R.C. / Tyssen, D.P. / Jardine, D.K. / Todd, B. / Passmore, M. / March, D.R. / Pattenden, L.K. / Bergman, D.A. / Alewood, D. / Hu, S.H. / Alewood, P.F. / Birch, C.J. / ...著者: Tyndall, J.D. / Reid, R.C. / Tyssen, D.P. / Jardine, D.K. / Todd, B. / Passmore, M. / March, D.R. / Pattenden, L.K. / Bergman, D.A. / Alewood, D. / Hu, S.H. / Alewood, P.F. / Birch, C.J. / Martin, J.L. / Fairlie, D.P. #1: ジャーナル: Biochemistry / 年: 1999タイトル: Molecular Recognition of Macrocyclic Peptidomimetic Inhibitors by HIV-1 Protease. 著者: Martin, J.L. / Begun, J. / Schindeler, A. / Wickramasinghe, W.A. / Alewood, D. / Alewood, P.F. / Bergman, D.A. / Brinkworth, R.I. / Abbenante, G. / March, D. / Reid, R.C. / Fairlie, D.P. #2: ジャーナル: J.Am.Chem.Soc. / 年: 1996タイトル: Substrate Based Cyclic Peptidomimetics Of Phe Ile Val That Inhibit HIV-1 Protease Using a Novel Enzyme Binding Mode. 著者: March, D. / Abbenante, G. / Bergman, D. / Brinkworth, R.I. / Wickramasinghe, W. / Begun, J. / Martin, J.L. / Fairlie, D.P. #3: ジャーナル: J.Am.Chem.Soc. / 年: 1995タイトル: Regioselective Structural and Functional Mimicry Of Peptides: Design Of Hydrolytically Stable Cyclic Peptidomimetic Inhibitors Of HIV-1 Protease. 著者: Abbenante, G. / March, D. / Bergman, D. / Hunt, P.A. / Garnham, B. / Dancer, R.J. / Martin, J.L. / Fairlie, D.P. | ||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 1d4l.cif.gz | 52.2 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb1d4l.ent.gz | 41 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 1d4l.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| 文書・要旨 | 1d4l_validation.pdf.gz | 1 MB | 表示 | wwPDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | 1d4l_full_validation.pdf.gz | 1 MB | 表示 | |
| XML形式データ | 1d4l_validation.xml.gz | 11.7 KB | 表示 | |
| CIF形式データ | 1d4l_validation.cif.gz | 15.9 KB | 表示 | |
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/d4/1d4lftp://data.pdbj.org/pub/pdb/validation_reports/d4/1d4l | HTTPS FTP |
-関連構造データ











-リンク
 PDBj
PDBj

- 集合体
集合体
| 登録構造単位 | 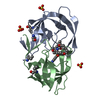
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| 単位格子 |
|
-要素
| #1: タンパク質 | 分子量: 10765.687 Da / 分子数: 2 / 変異: Q7K, L33I, C67(ABA), C95(ABA) / 由来タイプ: 合成 詳細: SF2 isolate, chemically synthesised protein corresponds to the protease from HIV-1, with 4 mutations per monomer 参照: UniProt: P03369, HIV-1 retropepsin #2: 化合物 |   分子量: 96.063 Da / 分子数: 3 / 由来タイプ: 合成 / 式: SO4 分子量: 96.063 Da / 分子数: 3 / 由来タイプ: 合成 / 式: SO4#3: 化合物 | ChemComp-PI9 / ( | 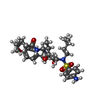  分子量: 616.812 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C32H48N4O6S 分子量: 616.812 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C32H48N4O6S#4: 水 | ChemComp-HOH / |  分子量: 18.015 Da / 分子数: 121 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 121 / 由来タイプ: 天然 / 式: H2O |
|---|
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 2.16 Å3/Da / 溶媒含有率: 43.18 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 結晶化 | 温度: 293 K / 手法: 蒸気拡散法, ハンギングドロップ法 / pH: 5.5 詳細: ammonium sulfate. acetate buffer, , pH 5.5, VAPOR DIFFUSION, HANGING DROP, temperature 293.0K | ||||||||||||||||||||||||||||||
| 結晶化 | *PLUS 温度: 20 ℃ / 詳細: inhibitor:protein molar ratio is 10:1 | ||||||||||||||||||||||||||||||
| 溶液の組成 | *PLUS
|
-データ収集
| 回折 | 平均測定温度: 298 K |
|---|---|
| 放射光源 | 由来: 回転陽極 / タイプ: RIGAKU RU200 / 波長: 1.54 |
| 検出器 | タイプ: RIGAKU RAXIS IIC / 検出器: IMAGE PLATE / 日付: 1998年5月8日 |
| 放射 | プロトコル: SINGLE WAVELENGTH / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray |
| 放射波長 | 波長: 1.54 Å / 相対比: 1 |
| 反射 | 解像度: 1.75→50 Å / Num. all: 62454 / Num. obs: 59019 / % possible obs: 94.5 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 1 / 冗長度: 3.38 % / Biso Wilson estimate: 10.2 Å2 / Rmerge(I) obs: 0.083 / Net I/σ(I): 9.5 |
| 反射 シェル | 解像度: 1.75→1.83 Å / 冗長度: 3.35 % / Rmerge(I) obs: 0.302 / Num. unique all: 1621 / % possible all: 84.6 |
| 反射 | *PLUS Num. obs: 18470 / Num. measured all: 62454 |
| 反射 シェル | *PLUS % possible obs: 84.6 % / Mean I/σ(I) obs: 2.1 |
- 解析
解析
| ソフトウェア |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 解像度: 1.75→8 Å / Rfactor Rfree error: 0.005 / Data cutoff high absF: 10000000 / Data cutoff low absF: 0.001 / Isotropic thermal model: RESTRAINED / 交差検証法: THROUGHOUT / σ(F): 0 / σ(I): 0 / 立体化学のターゲット値: Engh & Huber
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | Biso mean: 19.5 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 1.75→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS精密化 シェル | 解像度: 1.75→1.86 Å / Rfactor Rfree error: 0.02 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ソフトウェア | *PLUS 名称: X-PLOR / バージョン: 3.851 / 分類: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化 | *PLUS σ(F): 0 / % reflection Rfree: 9.8 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | *PLUS Biso mean: 19.5 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS精密化 シェル | *PLUS Rfactor Rfree: 0.32 / % reflection Rfree: 9.3 % / Rfactor Rwork: 0.281 / Rfactor obs: 0.288 |
