 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3bxr: Crystal Structures Of Highly Constrained Substrate And Hydrolysis... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3bxr | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structures Of Highly Constrained Substrate And Hydrolysis Products Bound To HIV-1 Protease. Implications For Catalytic Mechanism | ||||||
 Components Components | Protease | ||||||
 Keywords Keywords | HYDROLASE / HIV protease / HIVPR / substrate / product / AIDS / Aspartyl protease / Capsid maturation / Core protein / Cytoplasm / DNA integration / DNA recombination / DNA-directed DNA polymerase / Endonuclease / Lipoprotein / Magnesium / Membrane / Metal-binding / Multifunctional enzyme / Myristate / Nuclease / Nucleotidyltransferase / Nucleus / Phosphoprotein / RNA-binding / RNA-directed DNA polymerase / Transferase / Viral nucleoprotein / Virion / Zinc / Zinc-finger | ||||||
| Function / homology |  Function and homology information Function and homology informationHIV-1 retropepsin / symbiont-mediated activation of host apoptosis / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / DNA integration / viral genome integration into host DNA / establishment of integrated proviral latency / RNA-directed DNA polymerase / RNA stem-loop binding ...HIV-1 retropepsin / symbiont-mediated activation of host apoptosis / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / DNA integration / viral genome integration into host DNA / establishment of integrated proviral latency / RNA-directed DNA polymerase / RNA stem-loop binding / viral penetration into host nucleus / host multivesicular body / RNA-directed DNA polymerase activity / RNA-DNA hybrid ribonuclease activity / Transferases; Transferring phosphorus-containing groups; Nucleotidyltransferases / host cell / viral nucleocapsid / DNA recombination / DNA-directed DNA polymerase / aspartic-type endopeptidase activity / Hydrolases; Acting on ester bonds / DNA-directed DNA polymerase activity / symbiont-mediated suppression of host gene expression / viral translational frameshifting / symbiont entry into host cell / lipid binding / host cell nucleus / host cell plasma membrane / virion membrane / structural molecule activity / proteolysis / DNA binding / zinc ion binding Similarity search - Function | ||||||
| Method |  X-RAY DIFFRACTION / FOURIER SYNTHESIS / Resolution: 1.6 Å X-RAY DIFFRACTION / FOURIER SYNTHESIS / Resolution: 1.6 Å | ||||||
 Authors Authors | Tyndall, J.D. / Pattenden, L.K. / Reid, R.C. / Hu, S.H. / Alewood, D. / Alewood, P.F. / Walsh, T. / Fairlie, D.P. / Martin, J.L. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2008 Title: Crystal Structures of Highly Constrained Substrate and Hydrolysis Products Bound to HIV-1 Protease. Implications for the Catalytic Mechanism Authors: Tyndall, J.D. / Pattenden, L.K. / Reid, R.C. / Hu, S.H. / Alewood, D. / Alewood, P.F. / Walsh, T. / Fairlie, D.P. / Martin, J.L. #1: Journal: Biochemistry / Year: 1999Title: Molecular recognition of macrocyclic peptidomimetic inhibitors by HIV-1 protease Authors: Martin, J.L. / Begun, J. / Schindeler, A. / Wickramasinghe, W.A. / Alewood, D. / Alewood, P.F. / Bergman, D.A. / Brinkworth, R.I. / Abbenante, G. / March, D.R. / Reid, R.C. / Fairlie, D.P. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3bxr.cif.gz | 61.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3bxr.ent.gz | 42.8 KB | Display | PDB format |
| PDBx/mmJSON format | 3bxr.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bx/3bxrftp://data.pdbj.org/pub/pdb/validation_reports/bx/3bxr | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3bxsC  1cpiS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |

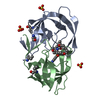









-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 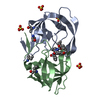
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 10764.701 Da / Num. of mol.: 2 / Fragment: UNP residues 491-589 Mutation: Q7K, D25N, L33I, C67(ABA), C95(ABA), Q107K, D125N, L133I, C167(ABA), C195(ABA) Source method: obtained synthetically Details: Chemically synthesized protein corresponding to the protease from the HIV1 References: UniProt: P03369, HIV-1 retropepsin #2: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4#3: Chemical | ChemComp-DRR / ( | 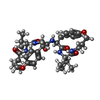  Mass: 677.830 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C37H51N5O7 Mass: 677.830 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C37H51N5O7#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 207 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 207 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.11 Å3/Da / Density % sol: 41.81 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 5.5 Details: Acetate, (NH4)2SO4, pH5.5, vapor diffusion, hanging drop, temperature 293K, VAPOR DIFFUSION, HANGING DROP |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: RIGAKU RAXIS IIC / Detector: IMAGE PLATE / Date: Apr 16, 1998 / Details: Yale mirrors | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Ni FILTER / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Rmerge(I) obs: 0.034 / Χ2: 1.02 / D res high: 1.6 Å / D res low: 50 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Diffraction reflection shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.6→50 Å / Num. all: 22699 / Num. obs: 22699 / % possible obs: 91.7 % / Observed criterion σ(I): 0 / Redundancy: 3.1 % / Rmerge(I) obs: 0.034 / Net I/σ(I): 22.5 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Resolution: 1.6→1.66 Å / Rmerge(I) obs: 0.093 / Mean I/σ(I) obs: 5.9 / Num. unique all: 1542 / % possible all: 64.1 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS Starting model: PDB ENTRY 1CPI Resolution: 1.6→8 Å / Cross valid method: THROUGHOUT / σ(F): 0
| |||||||||||||||||||||||||
| Solvent computation | Bsol: 88.063 Å2 | |||||||||||||||||||||||||
| Displacement parameters | Biso mean: 14.191 Å2
| |||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.17 Å | |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.6→8 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.6→1.61 Å
| |||||||||||||||||||||||||
| Xplor file |
|
