 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1a5v: ASV INTEGRASE CORE DOMAIN WITH HIV-1 INTEGRASE INHIBITOR Y3 AND M... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1a5v | ||||||
|---|---|---|---|---|---|---|---|
| Title | ASV INTEGRASE CORE DOMAIN WITH HIV-1 INTEGRASE INHIBITOR Y3 AND MN CATION | ||||||
 Components Components | INTEGRASE | ||||||
 Keywords Keywords | HYDROLASE / ENDONUCLEASE / HIV-1 INTEGRASE INHIBITOR | ||||||
| Function / homology |  Function and homology information Function and homology informationHydrolases; Acting on peptide bonds (peptidases); Aspartic endopeptidases / ribonuclease H / DNA integration / viral genome integration into host DNA / establishment of integrated proviral latency / RNA-directed DNA polymerase / RNA stem-loop binding / virion component / RNA-directed DNA polymerase activity / RNA-DNA hybrid ribonuclease activity ...Hydrolases; Acting on peptide bonds (peptidases); Aspartic endopeptidases / ribonuclease H / DNA integration / viral genome integration into host DNA / establishment of integrated proviral latency / RNA-directed DNA polymerase / RNA stem-loop binding / virion component / RNA-directed DNA polymerase activity / RNA-DNA hybrid ribonuclease activity / Transferases; Transferring phosphorus-containing groups; Nucleotidyltransferases / viral nucleocapsid / DNA recombination / DNA-directed DNA polymerase / aspartic-type endopeptidase activity / Hydrolases; Acting on ester bonds / DNA-directed DNA polymerase activity / viral translational frameshifting / symbiont entry into host cell / proteolysis / DNA binding / zinc ion binding Similarity search - Function | ||||||
| Biological species |  Rous sarcoma virus Rous sarcoma virus | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.9 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | ||||||
 Authors Authors | Lubkowski, J. / Yang, F. / Alexandratos, J. / Wlodawer, A. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 1998 Title: Structure of the catalytic domain of avian sarcoma virus integrase with a bound HIV-1 integrase-targeted inhibitor. Authors: Lubkowski, J. / Yang, F. / Alexandratos, J. / Wlodawer, A. / Zhao, H. / Burke Jr., T.R. / Neamati, N. / Pommier, Y. / Merkel, G. / Skalka, A.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1a5v.cif.gz | 46.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1a5v.ent.gz | 31.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1a5v.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/a5/1a5vftp://data.pdbj.org/pub/pdb/validation_reports/a5/1a5v | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1a5wC  1a5xC  1asvS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | THE AUTHOR MAINTAINS THAT THE BIOLOGICAL UNIT IS NOT YET KNOWN. THE MINIMUM MULTIMER IS BELIEVED TO CONTAIN AT LEAST THE DIMER GENERATED BT THE TRANSFORMATION IN REMARK 350, SHOWN IN BOTH HIV-1 AND ASV INTEGRASE CORE DOMAIN STRUCTURES. |
-Components
| #1: Protein | Mass: 17398.922 Da / Num. of mol.: 1 / Fragment: CATALYTIC CORE DOMAIN Source method: isolated from a genetically manipulated source Details: P03354 FRAGMENT OF POLYPROTEIN POL-RSVP Source: (gene. exp.) Rous sarcoma virus (strain Schmidt-Ruppin)Genus: Alpharetrovirus / Species: Rous sarcoma virus / Strain: Schmidt-Ruppin / Description: SEE JRNL REFERENCE / Plasmid: PRC23IN(52-207) / Production host:  References: UniProt: P03354, UniProt: O92956*PLUS, RNA-directed DNA polymerase |
|---|---|
| #2: Chemical | ChemComp-MN /   Mass: 54.938 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mn |
| #3: Chemical | ChemComp-Y3 / 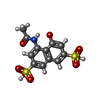  Mass: 361.348 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H11NO8S2 Mass: 361.348 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H11NO8S2 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 172 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 172 / Source method: isolated from a natural source / Formula: H2O |
| Sequence details | THE APPARENT DISCREPANCY BETWEEN THE SEQUENCE PRESENTED HERE AND THE "POL_RSVP" SEQUENCE IS A ...THE APPARENT DISCREPANC |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.52 Å3/Da / Density % sol: 38 % | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7.5 Details: 20% PEG 4000, 10% ISOPROPANOL, 0.1M NA HEPES, PH 7.5 | |||||||||||||||||||||||||
| Crystal grow | *PLUS Method: unknown | |||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 295 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: ENRAF-NONIUS FR591 / Wavelength: 1.5418 |
| Detector | Type: MAC Science DIP-2000 / Detector: IMAGE PLATE / Date: Aug 5, 1997 / Details: MIRRORS |
| Radiation | Monochromator: DOUBLE CRYSTAL SI(111) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→20 Å / Num. obs: 14653 / % possible obs: 98.2 % / Observed criterion σ(I): -3 / Redundancy: 6.16 % / Rsym value: 0.066 / Net I/σ(I): 16.5 |
| Reflection shell | Resolution: 1.9→1.97 Å / Rsym value: 0.354 / % possible all: 98.1 |
| Reflection | *PLUS Num. measured all: 92081 / Rmerge(I) obs: 0.066 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1ASV Resolution: 1.9→8 Å / Data cutoff high absF: 100000 / Data cutoff low absF: 0.1 / Cross valid method: FREE R / σ(F): 2 Details: ATOMIC OCCUPANCIES OF DISORDERED ATOMS ARE SET TO 0.00 IN THE COORDINATES,
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 29.65 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.9→1.99 Å / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
