 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-22531: The Cryo-EM structure of the Glutamate decarboxylase from Escheri... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-22531 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | The Cryo-EM structure of the Glutamate decarboxylase from Escherichia coli | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Glutamate decarboxylase beta / OXIDOREDUCTASE / LYASE | |||||||||
| Function / homology |  Function and homology information Function and homology informationglutamate decarboxylase / glutamate decarboxylase activity / intracellular pH elevation / L-glutamate catabolic process / pyridoxal phosphate binding / cytosol Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.59 Å | |||||||||
 Authors Authors | Su C-C | |||||||||
| Funding support |  United States, 1 items United States, 1 items
| |||||||||
 Citation Citation | Journal: Nat Methods / Year: 2021 Title: A 'Build and Retrieve' methodology to simultaneously solve cryo-EM structures of membrane proteins. Authors: Chih-Chia Su / Meinan Lyu / Christopher E Morgan / Jani Reddy Bolla / Carol V Robinson / Edward W Yu /  Abstract: Single-particle cryo-electron microscopy (cryo-EM) has become a powerful technique in the field of structural biology. However, the inability to reliably produce pure, homogeneous membrane protein ...Single-particle cryo-electron microscopy (cryo-EM) has become a powerful technique in the field of structural biology. However, the inability to reliably produce pure, homogeneous membrane protein samples hampers the progress of their structural determination. Here, we develop a bottom-up iterative method, Build and Retrieve (BaR), that enables the identification and determination of cryo-EM structures of a variety of inner and outer membrane proteins, including membrane protein complexes of different sizes and dimensions, from a heterogeneous, impure protein sample. We also use the BaR methodology to elucidate structural information from Escherichia coli K12 crude membrane and raw lysate. The findings demonstrate that it is possible to solve high-resolution structures of a number of relatively small (<100 kDa) and less abundant (<10%) unidentified membrane proteins within a single, heterogeneous sample. Importantly, these results highlight the potential of cryo-EM for systems structural proteomics. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_22531.map.gz | 6 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-22531-v30.xmlemd-22531.xml | 11.2 KB 11.2 KB | Display Display | EMDB header |
| Images | 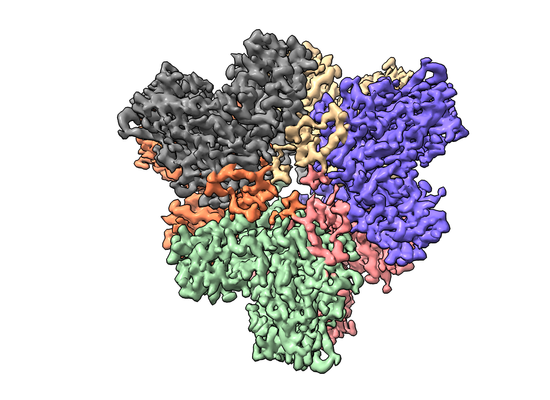 emd_22531.png emd_22531.png | 186.4 KB | ||
| Filedesc metadata | emd-22531.cif.gz | 5.4 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-22531ftp://ftp.pdbj.org/pub/emdb/structures/EMD-22531 http://ftp.pdbj.org/pub/emdb/structures/EMD-22531ftp://ftp.pdbj.org/pub/emdb/structures/EMD-22531 | HTTPS FTP |
-Related structure data
| Related structure data |  7jzhMC  6wtiC  6wtzC  6wu0C  6wu6C  7jz2C  7jz3C  7jz6C M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |













-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_22531.map.gz / Format: CCP4 / Size: 6.4 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & slices | Image control
Images are generated by Spider. generated in cubic-lattice coordinate | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.08 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
- Sample components
Sample components
-Entire : Glutamate decarboxylase
| Entire | Name: Glutamate decarboxylase |
|---|---|
| Components |
|
-Supramolecule #1: Glutamate decarboxylase
| Supramolecule | Name: Glutamate decarboxylase / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: |
-Macromolecule #1: Glutamate decarboxylase
| Macromolecule | Name: Glutamate decarboxylase / type: protein_or_peptide / ID: 1 / Number of copies: 6 / Enantiomer: LEVO / EC number: glutamate decarboxylase |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 52.727957 KDa |
| Sequence | String: MDKKQVTDLR SELLDSRFGA KSISTIAESK RFPLHEMRDD VAFQIINDEL YLDGNARQNL ATFCQTWDDE NVHKLMDLSI NKNWIDKEE YPQSAAIDLR CVNMVADLWH APAPKNGQAV GTNTIGSSEA CMLGGMAMKW RWRKRMEAAG KPTDKPNLVC G PVQICWHK ...String: MDKKQVTDLR SELLDSRFGA KSISTIAESK RFPLHEMRDD VAFQIINDEL YLDGNARQNL ATFCQTWDDE NVHKLMDLSI NKNWIDKEE YPQSAAIDLR CVNMVADLWH APAPKNGQAV GTNTIGSSEA CMLGGMAMKW RWRKRMEAAG KPTDKPNLVC G PVQICWHK FARYWDVELR EIPMRPGQLF MDPKRMIEAC DENTIGVVPT FGVTYTGNYE FPQPLHDALD KFQADTGIDI DM HIDAASG GFLAPFVAPD IVWDFRLPRV KSISASGHKF GLAPLGCGWV IWRDEEALPQ ELVFNVDYLG GQIGTFAINF SRP AGQVIA QYYEFLRLGR EGYTKVQNAS YQVAAYLADE IAKLGPYEFI CTGRPDEGIP AVCFKLKDGE DPGYTLYDLS ERLR LRGWQ VPAFTLGGEA TDIVVMRIMC RRGFEMDFAE LLLEDYKASL KYLSDHPKLQ GIAQQNSFKH T UniProtKB: Glutamate decarboxylase |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.5 mg/mL |
|---|---|
| Buffer | pH: 7.5 |
| Vitrification | Cryogen name: ETHANE |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K3 BIOQUANTUM (6k x 4k) / Average electron dose: 50.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
