 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-10948: Condensin complex from S.cerevisiae ATP-free apo non-engaged stat... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-10948 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
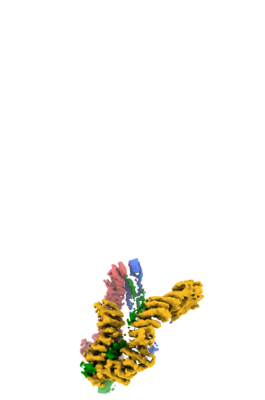
| Title | Condensin complex from S.cerevisiae ATP-free apo non-engaged state: focused refinement on head segment | ||||||||||||
 Map data Map data | |||||||||||||
 Sample Sample |
| ||||||||||||
| Biological species |  | ||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 4.2 Å | ||||||||||||
 Authors Authors | Lee B-G / Cawood C / Gutierrez-Escribano P / Nakane T / Merkel F / Hassler M / Haering CH / Aragon L / Lowe J | ||||||||||||
| Funding support |  United Kingdom, 3 items United Kingdom, 3 items
| ||||||||||||
 Citation Citation | Journal: Nat Struct Mol Biol / Year: 2020 Title: Cryo-EM structures of holo condensin reveal a subunit flip-flop mechanism. Authors: Byung-Gil Lee / Fabian Merkel / Matteo Allegretti / Markus Hassler / Christopher Cawood / Léa Lecomte / Francis J O'Reilly / Ludwig R Sinn / Pilar Gutierrez-Escribano / Marc Kschonsak / Sol ...Authors: Byung-Gil Lee / Fabian Merkel / Matteo Allegretti / Markus Hassler / Christopher Cawood / Léa Lecomte / Francis J O'Reilly / Ludwig R Sinn / Pilar Gutierrez-Escribano / Marc Kschonsak / Sol Bravo / Takanori Nakane / Juri Rappsilber / Luis Aragon / Martin Beck / Jan Löwe / Christian H Haering /   Abstract: Complexes containing a pair of structural maintenance of chromosomes (SMC) family proteins are fundamental for the three-dimensional (3D) organization of genomes in all domains of life. The ...Complexes containing a pair of structural maintenance of chromosomes (SMC) family proteins are fundamental for the three-dimensional (3D) organization of genomes in all domains of life. The eukaryotic SMC complexes cohesin and condensin are thought to fold interphase and mitotic chromosomes, respectively, into large loop domains, although the underlying molecular mechanisms have remained unknown. We used cryo-EM to investigate the nucleotide-driven reaction cycle of condensin from the budding yeast Saccharomyces cerevisiae. Our structures of the five-subunit condensin holo complex at different functional stages suggest that ATP binding induces the transition of the SMC coiled coils from a folded-rod conformation into a more open architecture. ATP binding simultaneously triggers the exchange of the two HEAT-repeat subunits bound to the SMC ATPase head domains. We propose that these steps result in the interconversion of DNA-binding sites in the catalytic core of condensin, forming the basis of the DNA translocation and loop-extrusion activities. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_10948.map.gz | 13.8 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-10948-v30.xmlemd-10948.xml | 13.4 KB 13.4 KB | Display Display | EMDB header |
| Images |  emd_10948.png emd_10948.png | 28.7 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-10948ftp://ftp.pdbj.org/pub/emdb/structures/EMD-10948 http://ftp.pdbj.org/pub/emdb/structures/EMD-10948ftp://ftp.pdbj.org/pub/emdb/structures/EMD-10948 | HTTPS FTP |
-Related structure data
| Related structure data |  6yvdC  6yvuC  6yvvC  10946 C: citing same article ( |
|---|---|
| Similar structure data |














-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_10948.map.gz / Format: CCP4 / Size: 209.3 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.2 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : Condensin
| Entire | Name: Condensin |
|---|---|
| Components |
|
-Supramolecule #1: Condensin
| Supramolecule | Name: Condensin / type: complex / ID: 1 / Parent: 0 Details: complex of 5 protein subunits: Smc2; Smc4; Brn1; Ycs4; Ycg1 |
|---|---|
| Source (natural) | Organism: |
| Recombinant expression | Organism: |
| Molecular weight | Theoretical: 500 MDa |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.2 mg/mL |
|---|---|
| Buffer | pH: 7.5 / Component - Name: Tris |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 100 K / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Specialist optics | Phase plate: VOLTA PHASE PLATE / Energy filter - Slit width: 20 eV |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Number grids imaged: 1 / Number real images: 10000 / Average exposure time: 10.0 sec. / Average electron dose: 45.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 50.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |





