 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4hxq: Crystal structure of human Arginase-1 complexed with inhibitor 14 -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4hxq | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of human Arginase-1 complexed with inhibitor 14 | ||||||
 Components Components | Arginase-1 | ||||||
 Keywords Keywords | Hydrolase/Hydrolase Inhibitor / metalloenzyme / alpha/beta fold / Hydrolase / Arginine metabolism / Manganese / Hydrolase-Hydrolase Inhibitor complex | ||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of neutrophil mediated killing of fungus / Urea cycle / negative regulation of T-helper 2 cell cytokine production / arginase / arginase activity / urea cycle / response to nematode / defense response to protozoan / negative regulation of activated T cell proliferation / negative regulation of type II interferon-mediated signaling pathway ...positive regulation of neutrophil mediated killing of fungus / Urea cycle / negative regulation of T-helper 2 cell cytokine production / arginase / arginase activity / urea cycle / response to nematode / defense response to protozoan / negative regulation of activated T cell proliferation / negative regulation of type II interferon-mediated signaling pathway / L-arginine catabolic process / negative regulation of T cell proliferation / specific granule lumen / azurophil granule lumen / manganese ion binding / adaptive immune response / innate immune response / Neutrophil degranulation / : / extracellular region / nucleus / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.45 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.45 Å | ||||||
 Authors Authors | Cousido-Siah, A. / Mitschler, A. / Ruiz, F.X. / Whitehouse, D.L. / Golebiowski, A. / Ji, M. / Zhang, M. / Beckett, P. / Sheeler, R. / Andreoli, M. ...Cousido-Siah, A. / Mitschler, A. / Ruiz, F.X. / Whitehouse, D.L. / Golebiowski, A. / Ji, M. / Zhang, M. / Beckett, P. / Sheeler, R. / Andreoli, M. / Conway, B. / Mahboubi, K. / Schroeter, H. / Van Zandt, M.C. / Podjarny, A. | ||||||
 Citation Citation | Journal: J.Med.Chem. / Year: 2013 Title: Discovery of (R)-2-Amino-6-borono-2-(2-(piperidin-1-yl)ethyl)hexanoic Acid and Congeners As Highly Potent Inhibitors of Human Arginases I and II for Treatment of Myocardial Reperfusion Injury. Authors: Van Zandt, M.C. / Whitehouse, D.L. / Golebiowski, A. / Ji, M.K. / Zhang, M. / Beckett, R.P. / Jagdmann, G.E. / Ryder, T.R. / Sheeler, R. / Andreoli, M. / Conway, B. / Mahboubi, K. / ...Authors: Van Zandt, M.C. / Whitehouse, D.L. / Golebiowski, A. / Ji, M.K. / Zhang, M. / Beckett, R.P. / Jagdmann, G.E. / Ryder, T.R. / Sheeler, R. / Andreoli, M. / Conway, B. / Mahboubi, K. / D'Angelo, G. / Mitschler, A. / Cousido-Siah, A. / Ruiz, F.X. / Howard, E.I. / Podjarny, A.D. / Schroeter, H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4hxq.cif.gz | 142 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4hxq.ent.gz | 107.5 KB | Display | PDB format |
| PDBx/mmJSON format | 4hxq.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hx/4hxqftp://data.pdbj.org/pub/pdb/validation_reports/hx/4hxq | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4hwwC  4hzeC  4i06C  1d3vS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |





















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||
| 2 | 
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
| |||||||||
| Details | biological assembly is a trimer generated from monomer A in the asymmetric unit by the operations: -x+y, -x, z-l; -y, x-y, z+1; or from monomer B in the asymmetric unit by the operations: -x+y, -x, z; -y, x-y, z |
-Components
| #1: Protein | Mass: 33923.840 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: ARG1 / Plasmid: pET24a / Production host:  #2: Chemical | 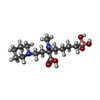  Mass: 317.209 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H30BN2O5 Mass: 317.209 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H30BN2O5#3: Chemical | ChemComp-MN /   Mass: 54.938 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mn#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 450 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 450 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.41 Å3/Da / Density % sol: 49.04 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 5 Details: 100 mM Malonic Acid, Imidazol, Boric system, 25% Polyethylene Glycol (PEG) 1500, vapor diffusion, hanging drop, temperature 277K |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X06SA / Wavelength: 0.70849 Å / Beamline: X06SA / Wavelength: 0.70849 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: PSI PILATUS 6M / Detector: PIXEL / Date: Oct 16, 2012 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: BARTELS MONOCHROMATOR / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.70849 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection twin | Operator: -h,-k,l / Fraction: 0.5 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.45→50 Å / Num. all: 111989 / Num. obs: 111989 / % possible obs: 99.5 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 4.9 % / Rmerge(I) obs: 0.053 / Χ2: 1.036 / Net I/σ(I): 9.3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: pdb entry 1D3V Resolution: 1.45→39.11 Å / Occupancy max: 1 / Occupancy min: 0.55 / Cross valid method: R-free / σ(F): 0 / Phase error: 18.33 / Stereochemistry target values: TWIN_LSQ_F
| ||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 25.94 Å2 / Biso mean: 9.656 Å2 / Biso min: 4.1 Å2 | ||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.45→39.11 Å
| ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.4496→1.4746 Å
|
