 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3kv2: HIGH RESOLUTION STRUCTURE OF HUMAN ARGINASE I IN COMPLEX WITH THE... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3kv2 | ||||||
|---|---|---|---|---|---|---|---|
| Title | HIGH RESOLUTION STRUCTURE OF HUMAN ARGINASE I IN COMPLEX WITH THE STRONG INHIBITOR N(omega)-hydroxy-nor-L-arginine (nor-NOHA) | ||||||
 Components Components | Arginase-1 | ||||||
 Keywords Keywords | HYDROLASE / STRONG INHIBITOR / nor-NOHA / ARGINASE / HIGH RESOLUTION / Alternative splicing / Arginine metabolism / Cytoplasm / Disease mutation / Manganese / Metal-binding / Phosphoprotein / Polymorphism / Urea cycle | ||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of neutrophil mediated killing of fungus / negative regulation of T-helper 2 cell cytokine production / Urea cycle / arginase / arginase activity / urea cycle / response to nematode / defense response to protozoan / negative regulation of activated T cell proliferation / negative regulation of type II interferon-mediated signaling pathway ...positive regulation of neutrophil mediated killing of fungus / negative regulation of T-helper 2 cell cytokine production / Urea cycle / arginase / arginase activity / urea cycle / response to nematode / defense response to protozoan / negative regulation of activated T cell proliferation / negative regulation of type II interferon-mediated signaling pathway / negative regulation of T cell proliferation / L-arginine catabolic process / specific granule lumen / azurophil granule lumen / manganese ion binding / adaptive immune response / innate immune response / Neutrophil degranulation / : / extracellular region / nucleus / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / PHASER / Resolution: 1.55 Å X-RAY DIFFRACTION / SYNCHROTRON / PHASER / Resolution: 1.55 Å | ||||||
 Authors Authors | Di Costanzo, L. / Christianson, D.W. | ||||||
 Citation Citation | Journal: Arch.Biochem.Biophys. / Year: 2010 Title: Inhibition of human arginase I by substrate and product analogues. Authors: Di Costanzo, L. / Ilies, M. / Thorn, K.J. / Christianson, D.W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3kv2.cif.gz | 136.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3kv2.ent.gz | 105.8 KB | Display | PDB format |
| PDBx/mmJSON format | 3kv2.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/kv/3kv2ftp://data.pdbj.org/pub/pdb/validation_reports/kv/3kv2 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3lp4C  3lp7C  2zavS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 34779.879 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: ARG1 / Production host:  #2: Chemical | ChemComp-MN /   Mass: 54.938 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mn#3: Chemical | 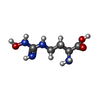  Type: L-peptide linking / Mass: 176.174 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C5H12N4O3 Type: L-peptide linking / Mass: 176.174 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C5H12N4O3#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 302 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 302 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.37 Å3/Da / Density % sol: 48.01 % |
|---|---|
| Crystal grow | Method: vapor diffusion, hanging drop Details: Hanging drops containing 3 L protein solution [3.5 mg/mL protein, 50 mM bicine (pH 8.5), 2 mM nor-NOHA, 100 M MnCl2] and 3 L precipitant solution [0.1 M bis-Tris (pH 6.5), 28% PEG monomethyl ...Details: Hanging drops containing 3 L protein solution [3.5 mg/mL protein, 50 mM bicine (pH 8.5), 2 mM nor-NOHA, 100 M MnCl2] and 3 L precipitant solution [0.1 M bis-Tris (pH 6.5), 28% PEG monomethyl ether 2000] were equilibrated over a 1 mL reservoir of precipitant solution at 2 C. , VAPOR DIFFUSION, HANGING DROP |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 24-ID-C / Wavelength: 1 Å / Beamline: 24-ID-C / Wavelength: 1 Å |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Oct 1, 2009 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.55→50 Å / Num. all: 90987 / Num. obs: 90987 / % possible obs: 98.2 % / Rmerge(I) obs: 0.144 / Rsym value: 0.144 / Net I/σ(I): 14.4 |
| Reflection shell | Resolution: 1.55→1.65 Å / Redundancy: 2 % / Rmerge(I) obs: 0.302 / % possible all: 96.2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: PHASER Starting model: 2ZAV monomer A Resolution: 1.55→50 Å Details: Data in the structure factor file is twinned. The operator is -h,-k,l, and the fraction is 0.5
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.55→50 Å
| ||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||
| LS refinement shell | Resolution: 1.55→1.61 Å /
|
