 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6q92: Crystal structure of human Arginase-1 at pH 7.0 in complex with ABH -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6q92 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of human Arginase-1 at pH 7.0 in complex with ABH | ||||||
 Components Components | Arginase-1 | ||||||
 Keywords Keywords | HYDROLASE / hydrolase inhibitor / borate / manganese cluster / pH-dependent | ||||||
| Function / homology |  Function and homology information Function and homology informationARG1 variants cause hyperargininemia / positive regulation of neutrophil mediated killing of fungus / negative regulation of T-helper 2 cell cytokine production / Urea cycle / arginase / arginase activity / urea cycle / defense response to protozoan / negative regulation of activated T cell proliferation / negative regulation of type II interferon-mediated signaling pathway ...ARG1 variants cause hyperargininemia / positive regulation of neutrophil mediated killing of fungus / negative regulation of T-helper 2 cell cytokine production / Urea cycle / arginase / arginase activity / urea cycle / defense response to protozoan / negative regulation of activated T cell proliferation / negative regulation of type II interferon-mediated signaling pathway / negative regulation of T cell proliferation / L-arginine catabolic process / specific granule lumen / azurophil granule lumen / manganese ion binding / adaptive immune response / innate immune response / Neutrophil degranulation / : / extracellular region / nucleus / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.5 Å | ||||||
 Authors Authors | Grobben, Y. / Uitdehaag, J.C.M. / Zaman, G.J.R. | ||||||
 Citation Citation | Journal: J Struct Biol X / Year: 2020 Title: Structural insights into human Arginase-1 pH dependence and its inhibition by the small molecule inhibitor CB-1158. Authors: Grobben, Y. / Uitdehaag, J.C.M. / Willemsen-Seegers, N. / Tabak, W.W.A. / de Man, J. / Buijsman, R.C. / Zaman, G.J.R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6q92.cif.gz | 150.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6q92.ent.gz | 114.9 KB | Display | PDB format |
| PDBx/mmJSON format | 6q92.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/q9/6q92ftp://data.pdbj.org/pub/pdb/validation_reports/q9/6q92 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6q9pC  6qafC  2aebS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||
| 2 | 
| ||||||||||||||||||
| Unit cell |
| ||||||||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 36951.227 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Details: N-terminal his tag with thrombin cleavage site / Source: (gene. exp.) Homo sapiens (human) / Gene: ARG1 / Plasmid: pET15b / Production host:  #2: Chemical | ChemComp-MN /   Mass: 54.938 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mn / Feature type: SUBJECT OF INVESTIGATION Mass: 54.938 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mn / Feature type: SUBJECT OF INVESTIGATION#3: Chemical | 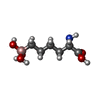  Mass: 191.998 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H15BNO5 / Feature type: SUBJECT OF INVESTIGATION Mass: 191.998 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H15BNO5 / Feature type: SUBJECT OF INVESTIGATION#4: Chemical |   Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na / Feature type: SUBJECT OF INVESTIGATION Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na / Feature type: SUBJECT OF INVESTIGATION#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 624 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 624 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.22 Å3/Da / Density % sol: 44.6 % / Description: clear rod-like hexagonal crystals |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 4 Details: Crystals were generated from 22 % PEG 1500, 0.2 M MIB buffer pH 4.0 (sodium malonate, imidazole and boric acid in a 2:3:3 molar ratio). Crystals were equilibrated to soaking solution (22 % ...Details: Crystals were generated from 22 % PEG 1500, 0.2 M MIB buffer pH 4.0 (sodium malonate, imidazole and boric acid in a 2:3:3 molar ratio). Crystals were equilibrated to soaking solution (22 % PEG 1500, 0.2 M MMT buffer pH 7.0 (DL-malic acid, MES, Tris base in a 1:2:2 molar ratio). Subsequently, crystals were gradually soaked for 13 days in soaking solution containing 15 mM ABH. Temp details: room temperature |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: MASSIF-1 / Wavelength: 0.966 Å / Beamline: MASSIF-1 / Wavelength: 0.966 Å | |||||||||||||||
| Detector | Type: DECTRIS PILATUS3 2M / Detector: PIXEL / Date: Sep 6, 2017 / Details: transfocator | |||||||||||||||
| Radiation | Monochromator: diamond beam splitter / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||
| Radiation wavelength | Wavelength: 0.966 Å / Relative weight: 1 | |||||||||||||||
| Reflection twin |
| |||||||||||||||
| Reflection | Resolution: 1.5→39.14 Å / Num. obs: 99464 / % possible obs: 98.2 % / Redundancy: 2.4 % / Biso Wilson estimate: 13.91 Å2 / CC1/2: 0.987 / Rmerge(I) obs: 0.097 / Rpim(I) all: 0.074 / Rrim(I) all: 0.122 / Χ2: 0.93 / Net I/σ(I): 6.9 | |||||||||||||||
| Reflection shell | Resolution: 1.5→1.53 Å / Redundancy: 2.1 % / Rmerge(I) obs: 0.608 / Mean I/σ(I) obs: 1.8 / Num. unique obs: 4790 / CC1/2: 0.293 / Rpim(I) all: 0.508 / Rrim(I) all: 0.796 / Χ2: 0.82 / % possible all: 96.4 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 2AEB Resolution: 1.5→39.14 Å / Cor.coef. Fo:Fc: 0.978 / Cor.coef. Fo:Fc free: 0.965 / SU B: 0.679 / SU ML: 0.026 / Cross valid method: THROUGHOUT / ESU R: 0.012 / ESU R Free: 0.011 Details: Hydrogens have been added in riding positions. Refinement with 'crystal is twinned' switched on. Refinement of anisotropic B-factors for the Manganese cluster only.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 16.294 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 1.5→39.14 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|