 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1hqf: CRYSTAL STRUCTURE OF THE BINUCLEAR MANGANESE METALLOENZYME ARGINA... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1hqf | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF THE BINUCLEAR MANGANESE METALLOENZYME ARGINASE COMPLEXED WITH N-HYDROXY-L-ARGININE | ||||||
 Components Components | ARGINASE 1 | ||||||
 Keywords Keywords | HYDROLASE / arginase / N-Hydroxy-L-Arginine (NOHA) / binuclear manganese cluster / metalloenzyme | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of L-arginine import across plasma membrane / Urea cycle / collagen biosynthetic process / mammary gland involution / positive regulation of neutrophil mediated killing of fungus / negative regulation of T-helper 2 cell cytokine production / arginine metabolic process / arginase / arginase activity / urea cycle ...regulation of L-arginine import across plasma membrane / Urea cycle / collagen biosynthetic process / mammary gland involution / positive regulation of neutrophil mediated killing of fungus / negative regulation of T-helper 2 cell cytokine production / arginine metabolic process / arginase / arginase activity / urea cycle / response to selenium ion / response to methylmercury / response to nematode / response to manganese ion / response to steroid hormone / response to vitamin A / response to amine / response to herbicide / Neutrophil degranulation / response to zinc ion / response to vitamin E / defense response to protozoan / negative regulation of activated T cell proliferation / negative regulation of type II interferon-mediated signaling pathway / response to amino acid / maternal process involved in female pregnancy / cellular response to dexamethasone stimulus / response to axon injury / response to cadmium ion / cellular response to transforming growth factor beta stimulus / negative regulation of T cell proliferation / cellular response to interleukin-4 / positive regulation of endothelial cell proliferation / lung development / cellular response to glucagon stimulus / liver development / female pregnancy / response to peptide hormone / cellular response to hydrogen peroxide / manganese ion binding / cellular response to lipopolysaccharide / response to lipopolysaccharide / adaptive immune response / response to xenobiotic stimulus / innate immune response / neuronal cell body / : / identical protein binding / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.9 Å | ||||||
 Authors Authors | Cox, J.D. / Cama, E. / Colleluori, D.M. / Ash, D.E. / Christianson, D.W. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2001 Title: Mechanistic and metabolic inferences from the binding of substrate analogues and products to arginase. Authors: Cox, J.D. / Cama, E. / Colleluori, D.M. / Pethe, S. / Boucher, J.L. / Mansuy, D. / Ash, D.E. / Christianson, D.W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1hqf.cif.gz | 187.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1hqf.ent.gz | 150 KB | Display | PDB format |
| PDBx/mmJSON format | 1hqf.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hq/1hqfftp://data.pdbj.org/pub/pdb/validation_reports/hq/1hqf | HTTPS FTP |
|---|
-Related structure data























-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | the biological assembly is a trimer |
-Components
| #1: Protein | Mass: 35019.098 Da / Num. of mol.: 3 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  #2: Chemical | ChemComp-MN /   Mass: 54.938 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Mn#3: Chemical | 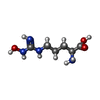  Type: L-peptide linking / Mass: 190.200 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C6H14N4O3 Type: L-peptide linking / Mass: 190.200 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C6H14N4O3#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 15 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 15 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.38 Å3/Da / Density % sol: 48.35 % | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 8.5 Details: PEG 8000, BICINE, manganese chloride, pH 8.5, VAPOR DIFFUSION, HANGING DROP, temperature 277K | ||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal | *PLUS Density % sol: 48 % | ||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / Details: Kanyo, Z.F., (1992) J.Mol.Biol., 224, 1175. | ||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL7-1 / Wavelength: 1.08 Å / Beamline: BL7-1 / Wavelength: 1.08 Å |
| Detector | Type: MAR scanner 345 mm plate / Detector: IMAGE PLATE / Date: Jun 20, 2000 |
| Radiation | Monochromator: filter / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.08 Å / Relative weight: 1 |
| Reflection | Resolution: 2.9→30 Å / Num. all: 17524 / Num. obs: 17424 / % possible obs: 83.7 % / Observed criterion σ(F): 2 / Observed criterion σ(I): 2 / Redundancy: 1.8 % / Rmerge(I) obs: 0.064 / Net I/σ(I): 11.4 |
| Reflection shell | Resolution: 2.9→3 Å / Redundancy: 1.8 % / Rmerge(I) obs: 0.302 / % possible all: 86.7 |
| Reflection | *PLUS Num. obs: 17524 / Num. measured all: 31263 |
| Reflection shell | *PLUS % possible obs: 96.7 % / Num. unique obs: 781 |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.9→30 Å / σ(F): 2 / σ(I): 2 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.9→30 Å
| ||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||
| Software | *PLUS Name: CNS / Classification: refinement | ||||||||||||||||||||
| Refine LS restraints | *PLUS
|
