 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1sux: CRYSTALLOGRAPHIC ANALYSIS OF THE COMPLEX BETWEEN TRIOSEPHOSPHATE ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1sux | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTALLOGRAPHIC ANALYSIS OF THE COMPLEX BETWEEN TRIOSEPHOSPHATE ISOMERASE FROM TRYPANOSOMA CRUZI AND 3-(2-benzothiazolylthio)-1-propanesulfonic acid | ||||||
 Components Components | Triosephosphate isomerase, glycosomal | ||||||
 Keywords Keywords | ISOMERASE / TRIOSEPHOSPHATE ISOMERASE / TRYPANOSOMA CRUZI / PROTEIN INTERFACES / BENZOTHIAZOLE INHIBITOR | ||||||
| Function / homology |  Function and homology information Function and homology informationglycosome / triose-phosphate isomerase / triose-phosphate isomerase activity / glyceraldehyde-3-phosphate biosynthetic process / glycerol catabolic process / glycolytic process / gluconeogenesis / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Tellez-Valencia, A. / Olivares-Illana, V. / Hernandez-Santoyo, A. / Perez-Montfort, R. / Costas, M. / Rodriguez-Romero, A. / Tuena De Gomez-Puyou, M. / Gomez-Puyou, A. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2004 Title: Inactivation of triosephosphate isomerase from Trypanosoma cruzi by an agent that perturbs its dimer interface. Authors: Tellez-Valencia, A. / Olivares-Illana, V. / Hernandez-Santoyo, A. / Perez-Montfort, R. / Costas, M. / Rodriguez-Romero, A. / Lopez-Calahorra, F. / Tuena De Gomez-Puyou, M. / Gomez-Puyou, A. #1: Journal: J.Mol.Biol. / Year: 1998Title: Differences in the intersubunit contacts in triosephosphate isomerase from two closely related pathogenic trypanosomes Authors: Maldonado, E. / Soriano-Garcia, M. / Moreno, A. / Cabrera, N. / Garza-Ramos, G. / De Gomez-Puyou, M. / Gomez-Puyou, A. / Perez-Montfort, R. #2: Journal: Proc.Natl.Acad.Sci.USA / Year: 1999Title: Crystal structure of triosephosphate isomerase from Trypanosoma cruzi in hexane Authors: Gao, X.G. / Maldonado, E. / Perez-Monfort, R. / Garza-Ramos, G. / De Gomez-Puyou, M.T. / Gomez-Puyou, A. / Rodriguez-Romero, A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1sux.cif.gz | 115.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1sux.ent.gz | 89.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1sux.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/su/1suxftp://data.pdbj.org/pub/pdb/validation_reports/su/1sux | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1tcdS S: Starting model for refinement |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 27360.438 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  #2: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: SO4#3: Chemical | ChemComp-BTS / | 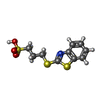  Mass: 289.394 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H11NO3S3 Mass: 289.394 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H11NO3S3#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 374 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 374 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.18 Å3/Da / Density % sol: 43.6 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: PEG 400, HEPES, ammonium sulfate, pH 7.5, VAPOR DIFFUSION, HANGING DROP, temperature 291K |
-Data collection
| Diffraction | Mean temperature: 113 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU / Wavelength: 1.54 Å |
| Detector | Type: RIGAKU RAXIS II / Detector: IMAGE PLATE / Date: May 21, 2002 / Details: mirrors |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.54 Å / Relative weight: 1 |
| Reflection | Resolution: 2→25 Å / Num. obs: 31622 / Observed criterion σ(F): 2 / Observed criterion σ(I): 2 / Redundancy: 2.5 % / Biso Wilson estimate: 22.7 Å2 / Rsym value: 0.04 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1TCD Resolution: 2→23.03 Å / Rfactor Rfree error: 0.004 / Data cutoff high absF: 1724206.77 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 Details: FIVE DISCRETELY DISORDERED RESIDUES: GLU 19, GLU 27, LYS 53, SER 97 OF MONOMER A AND SER 2 0F MONOMER B. RESIDUE MET 1 OF BOTH CHAINS AND SIDE CHAINS FROM CG OF RESIDUES LYS A 157, LYS A ...Details: FIVE DISCRETELY DISORDERED RESIDUES: GLU 19, GLU 27, LYS 53, SER 97 OF MONOMER A AND SER 2 0F MONOMER B. RESIDUE MET 1 OF BOTH CHAINS AND SIDE CHAINS FROM CG OF RESIDUES LYS A 157, LYS A 177, GLN A 182 AND LYS B 218 HAVE WEAK ELECTRON DENSITY AND ARE NOT INCLUDED IN THE MODEL.
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 59.7691 Å2 / ksol: 0.402909 e/Å3 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 19.5 Å2
| ||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→23.03 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.13 Å / Rfactor Rfree error: 0.01 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||
| Xplor file |
|
