

SeSAW Tutorial
In this tutorial, we provide explanation about the SeSAW, one of the service of PDBj.
See also the SeSAW documentation about basic things such as used technique.
- Submitting PDB entries to SeSAW
- Submitting structures to SeSAW
- Experimental structure examples
- Structural model examples
[Submitting PDB entries to SeSAW]
The SeSAW top page is shown below with forms needed for submitting a PDB entry filled out.

The email address is optional. You could also select to have the results sent to your web browser.
Points to keep in mind are:
- The chain ID is required: please confirm if you are not sure of the chain ID
- Only valid PDB entries will be processed so it is a good idea to check that your PDB ID exists
- Some queries can take up to an hour to complete so please be patient
- If your query is not completed in this time, or it is completed but the results seem strange, please send us an email at sesaw@protein.osaka-u.ac.jp
[Submitting structures to SeSAW]
Experimental structures
The SeSAW top page is shown below with forms needed for submitting a PDB-formatted file entry filled out.
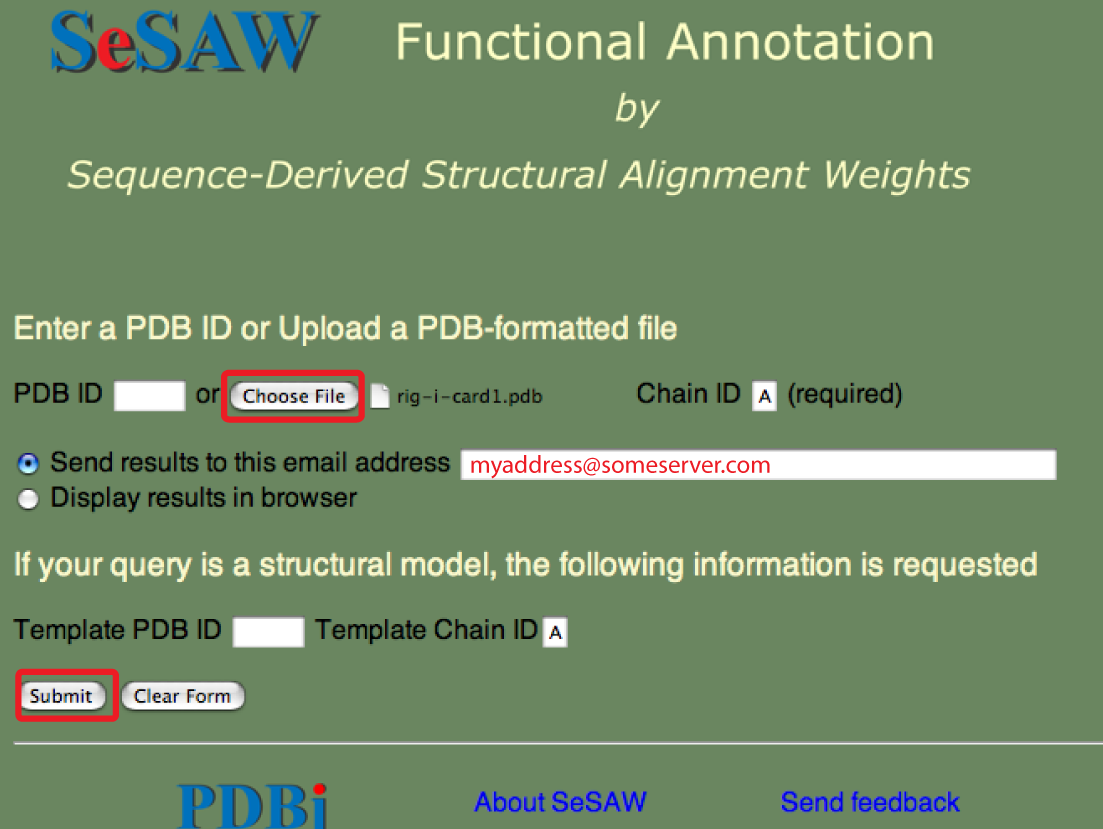
As, usual the email address is optional. You could also select to have the results sent to your web browser.
Points to keep in mind are the same as above except that the file-based queries tend to take longer because less of the steps have been pre-processed.
Structural models
Here the procedure is the same, but we recommend that the PDB ID of the template be included. This is because the structural neighbors of the template can then be looked up and retrieved quickly. This potentially represents a significant decrease in the time for the query.
[Experimental structure examples]
2czl
2czl is a structural genomics (SG) target solved as part of the Protein 3000 project. The steps for performing a SeSAW query using 2czl are as follows:
1. Input the PDB ID (2czl), chain ID (A), and email address as described above.
2. After a while, an email will be sent to you like the following:
From: sesaw@protein.osaka-u.ac.jp Subject: SeSAW Query 2czlA Date: October 7, 2008 5:48:15 PM JST To: xxxx Thank you for using the SeSAW functional annotation server. The results of your query will be stored for 2 weeks at the following URL: http://pdbjc01.protein.osaka-u.ac.jp:8888/tmp/SS17645/2czlA_top.html We hope these results will be useful for your research. Best wishes, -SeSAW
If you don't get an email after an hour or two, then something is probably wrong and you should send us an email. You can use the sesaw@protein.osaka-u.ac.jp address.
3. Follow the link in the email where a page like the one below should appear

Note that there are links for 2czlA (the whole chain) as well as the two domains. SeSAW always tries to break the query into domains, if possible, since this can dramatically affect the score.
4. Select the whole chain. A 'results table' should appear similar to the one shown below.
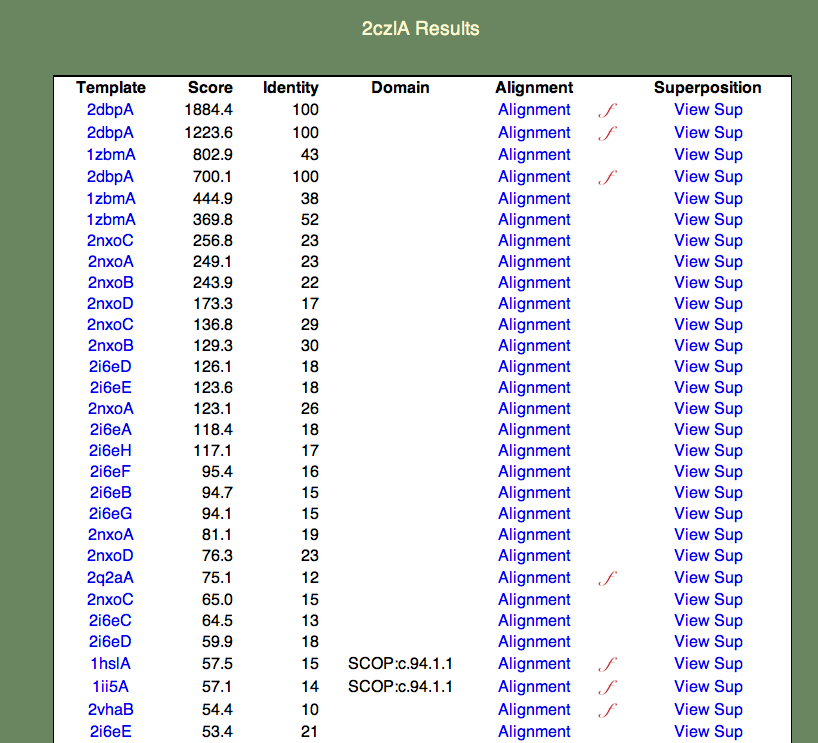
The format of this table is as follows:
- The Template column contains links to the PDBj Mine pages for PDB entries that are similar to 2czl in order of decreasing similarity. Note that a given entry may appear multiple times. This is because the templates are also broken into domains; for simplicity the domain numbers are not included in the results table.
- The Score column contains the raw SeSAW score.
- The Identity column contains the sequence identity of aligned residue pairs (i.e., gaps are not included).
- The Alignment column contains a link to the annotated Jalview alignment. If a red, cursive f appears to the right of the alignment link, functional annotations for this alignment exist.
- The Superposition column contains a link to an ASH structural superposition rendered by the jV molecular viewer.
5. Next we will get a general idea of the function of 2czl. If you go down the Template column, starting from the top, you will find that the top 25 or so templates are all SG targets. That is, they lack a detailed functional annotation, and, in most cases, have no primary literature reference. If you keep checking further down the list, however, you will find that the top templates with literature references are all amino acid binding proteins.
6. We will next examine a particular query-template alignment. Find the line containing the template 1ii5A. If you click on the alignment link, a page similar to the one below should appear.
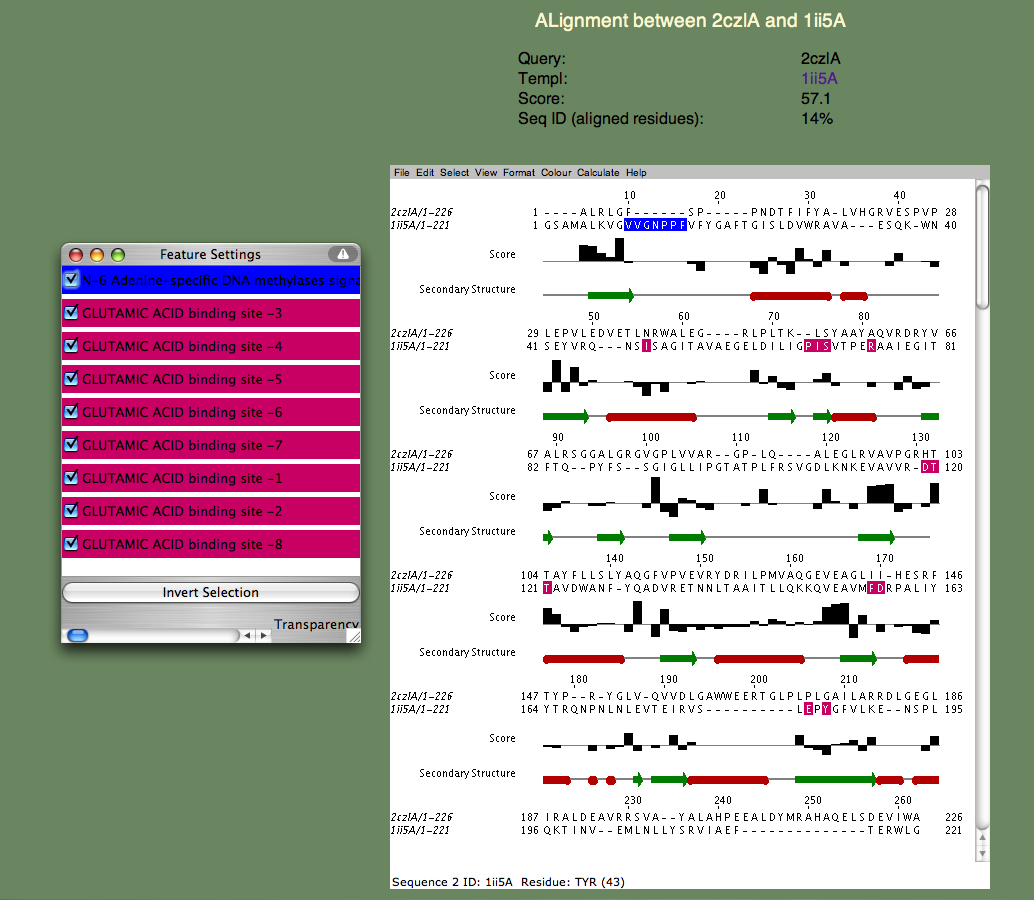
If the page does not appear, but instead a window appears with an 'X' in it, there is a problem running the Jalview applet on your browser. Please refer to the Jalview homepage for help. In particular, check their 'News Mailing List.' The Jalview people are very helpful and quick to respond to questions about their software. The alignment window gives us a much more detailed annotation of the query. Note the 'Features' window that pops up, listing all the continuous sequence motifs that have been functionally annotated. The first one is an 'N-6 Adenine-specific DNA methylases signature,' colored blue. Note that this sequence segment in the template corresponds to a gap in the alignment. In other words, 2czlA does not have the feature in question.
Next, lets look at the remaining features, which all correspond to a 'GLUTAMIC ACID binding site' and are colored pinkish purple. Note that these residues are spread throughout the sequence and most of them (all but a proline) do not align with gaps. Moreover, you can see that the bar-graph below the residues, indicating the per-residue SeSAW score, is favorable for three of these residues (S57,T106-T107 in the query) but is negative for others. This suggests that the location of the binding site probably corresponds to a related but different functional site in 2czl. Interestingly, selecting the template 2q2aA, which scores higher than 1ii5A, and binds a more basic amino acid (arginine) show that the same two threonines (T106-107) are conserved, but not S57. remaining residues differ. From these considerations, the likely location of the binding site, but not the exact ligand, can be predicted.
In fact, the 2czl structure was co-crystalized with tartaric acid. So, we can look at the location of the tartaric acid and find that it matches that of the amino acids in 1ii5A and 2q2aA. To see this, click the 2dbpA template (first on the list). Thus we are rather confident about the location of potential ligands, and we know that these ligands include tartaric acid. However, we can not say at this time what the function of 2czlA is.
7. We will next look at the structural superposition feature. If you click the 'View Sup' link on the 1ii5 line, a new page should appear containing a jV window. The first time you click this link, you will be prompted by the pop-up window shown below
which you can safely accept. The structural superposition should then appear as shown below
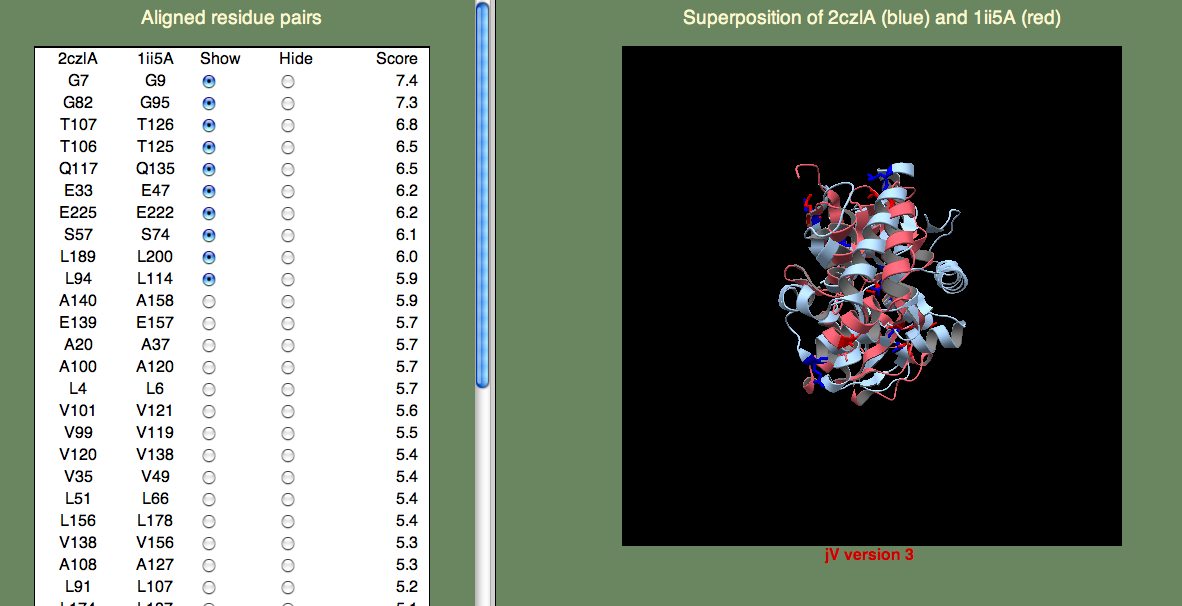
The panel on the left allows you to show or hide selected residue pairs. Again, these pairs are sorted, by the per-residue SeSAW score. By default, the top-10 residues are selected.
If we look for the conserved threonines (106-107) and serine (57), we can see they indeed form a cluster. Do any of the other top-scoring residue pairs belong in this cluster? You may have to click on quite as few residue pairs to find them, but A108 is also part of this cluster. Now, what about G82? This residue is rather far away, but it turns out to be important for opening the binding site to allow ligand access. So, we are happy to see this residue pair aligned with such a high score.
You can try to find other clusters by selecting different residue pairs. For example, the cluster formed by L4, L94, V99, A100, V101, Q117, V120, and E139 is located on the surface of one of the domains. It is tempting to think that this might have significance, and this could be further explored by examining the individual domains (2czlA_Dom-2 in this case).
1wjk
Here we will proceed directly to the analysis of the top-scoring alignments. The results table is shown below.
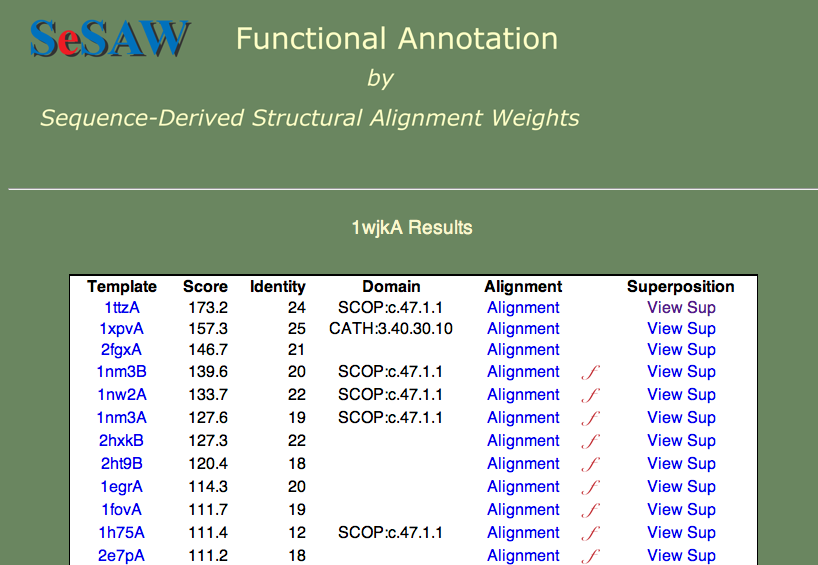
The two questions we want to answer are: 1) What is the function of 1wjk? and 2) What residues are likely to be functionally important? To answer the first question, what are the functions of the top-scoring templates? We find that they vary from unknown (1ttza,1xpvA,2fgxA) to glutaredoxin (1nm3B) to thioredoxin (1nw2A). Since these proteins are all part of the same superfamily, we can say that our protein probably belongs to this general group.
Next, to answer the second question, we have to know what Thioredoxins and Glutaredoxins do. Both are involved in signaling via oxidation/reduction (redox) reactions facilitated by their Cystine pair that can be either in the oxidized (unpaired) or reduced (disulphide bridge) state. Thioredoxins and Glutaredoxins differ in terms of what molecules they act on. Thioredoxin is reduced by Thioredoxin reductase, a larger protein. Glutaredoxin is reduced by glutathione, a smaller molecule. So, we want to find the cystine pair to identify the redox center. Then we want to look for possible protein-protein interaction sites (Glutaredoxin) or small molecule binding sites (Thioredoxin). I think you can easily find the cystines, since they score highest in the alignment. Next, lets consider what other residues score well. If we consider the highest-scoring (un-annotated) template 1ttzA, we find that there are a number of hydrophobic residues that score well (see below).
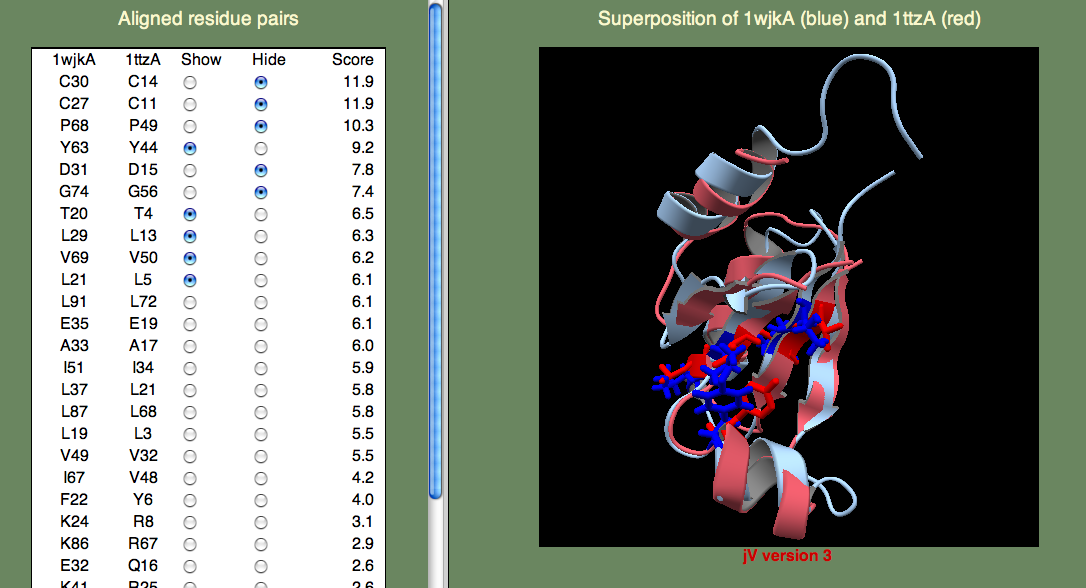
It could be that these residues are just part of the hydrophobic core that stabilizes the fold. Or, perhaps they are part of a protein-protein binding site? We can not be sure from just these data, but since the superfamily representing these proteins is growing rapidly through structural genomics efforts, I think that in the near future we will begin to learn more about their function.
1wj5
Here we we again would like to know the general function of the protein and identify potentially important residues. The top-scoring hits are shown below.
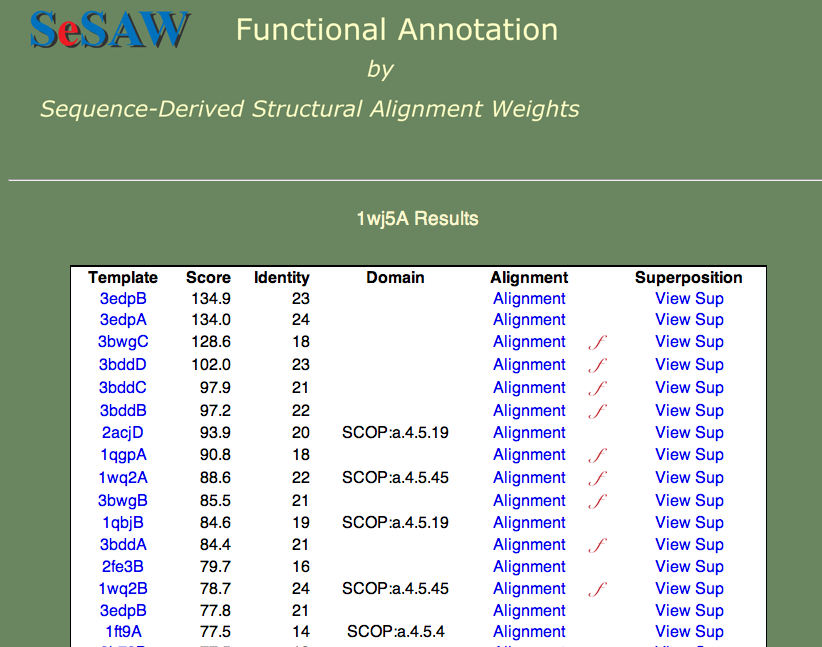
Can you guess the basic function? Again, this is a hard cases because we don't know the answer, but we can look at each of the templates and establish right away that nucleotide binding is a theme. Furthermore, if we check t alignment to the first annotated template (3bwg), shown below, we ask ourselves if the functional sites on 1wj5 match those on the template.
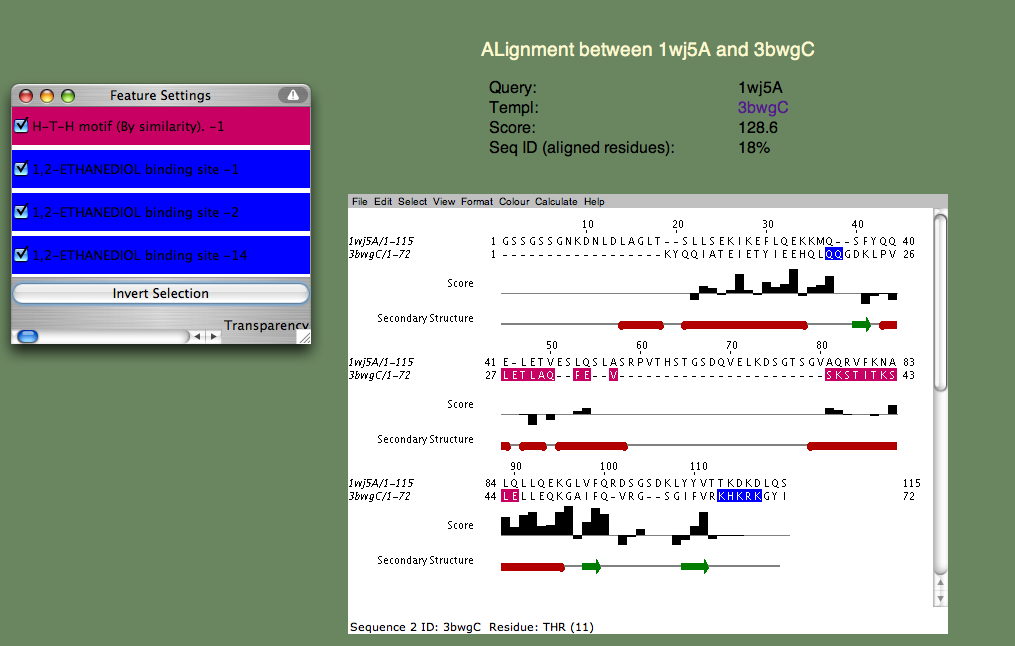
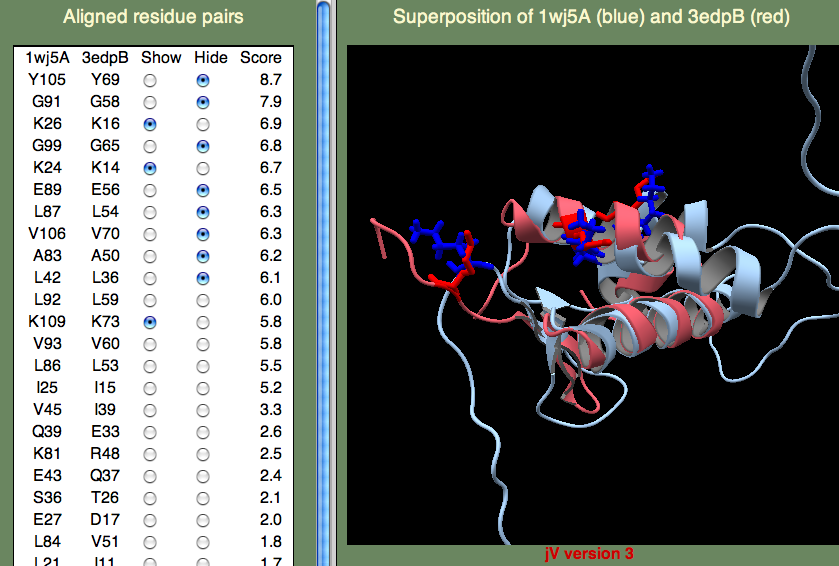
The H-T-H motif stands for Helix-Turn-Helix, and, as the name suggests, this is more of a structural motif than a sequence motif. Moreover, the sequence alignments in this (pink) region is rather poor. Therefore, it seem unlikely that 1wj5 and 3bwg bind nucleotides in the same manner. The second motif (1,2-ETHANEDIOL binding site) is the opposite: It doesn't align so well structurally, but there are a number of sequence similarities (Q35 and the Lysine residues at the C-terminus). These residue, along with the unaligned segment 'SRPVTHSTGSDQVELKDSGTSGV' make up rather disordered parts of 1wj5. It is known that disordered regions are often involved in transcriptional regulation, so it is tempting to think that this might be the case here. Perhaps the best clue as to where 1wj5 is likely to be involved in nucleotide interactions comes from examining the highest-scoring, unannotated, template 3edpB. The superposition shows three basic residues (K26,K24,and K109) score well and are somewhat clustered together. This is still very tentative, but it is probably our best guess at a nucleotide binding site.
[Structural model examples]
Helicase domain from RIG-I
1. First you have to build a model. This is an art in itself, so we will not cover it in any depth here. There are excellent resources such as SWISS-MODEL that offer information about model building. In this example, the HHpred server was used to build a model of the helicase domain in the Retinoic acid-inducible gene-I (RIG-I). First the whole sequence for mouse RIG-I was downloaded from UniProt by typing in the keywords 'mouse' and 'rig-i', clicking the accession ID, and then selecting FASTA at the upper-right part of the summary page. The FASTA sequence should look like:
>sp|Q6Q899|DDX58_MOUSE Probable ATP-dependent RNA helicase DDX58 OS=Mus musculus GN=Ddx58 PE=2 SV=1 MTAAQRQNLQAFRDYIKKILDPTYILSYMSSWLEDEEVQYIQAEKNNKGPMEAASLFLQY LLKLQSEGWFQAFLDALYHAGYCGLCEAIESWDFQKIEKLEEHRLLLRRLEPEFKATVDP NDILSELSECLINQECEEIRQIRDTKGRMAGAEKMAECLIRSDKENWPKVLQLALEKDNS KFSELWIVDKGFKRAESKADEDDGAEASSIQIFIQEEPECQNLSQNPGPPSEASSNNLHS PLKPRNYQLELALPAKKGKNTIICAPTGCGKTFVSLLICEHRLKKFPCGQKGKVVFFANQ IPVYEQQATVFSRYFERLGYNIASISGATSDSVSVQHIIEDNDIIILTPQILVNNLNNGA IPSLSVFTLMIFDECHNTSKNHPYNQIMFRYLDHKLGESRDPLPQVVGLTASVGVGDAKT AEEAMQHICKLCAALDASVIATVRDNVAELEQVVYKPQKISRKVASRTSNTFKCIISQLM KETEKLAKDVSEELGKLFQIQNREFGTQKYEQWIVGVHKACSVFQMADKEEESRVCKALF LYTSHLRKYNDALIISEDAQMTDALNYLKAFFHDVREAAFDETERELTRRFEEKLEELEK VSRGPSNENPKLRDLYLVLQEEYHLKPETKTILFVKTRALVDALKKWIEENPALSFLKPG ILTGRGRTNRATGMTLPAQKCVLEAFRASGDNNILIATSVADEGIDIAECNLVILYEYVG NVIKMIQTRGRGRARDSKCFLLTSSADVIEKEKANMIKEKIMNESILRLQTWDEMKFGKT VHRIQVNEKLLRDSQHKPQPVPDKENKKLLCGKCKNFACYTADIRVVETSHYTVLGDAFK ERFVCKPHPKPKIYDNFEKKAKIFCAKQNCSHDWGIFVRYKTFEIPVIKIESFVVEDIVS GVQNRHSKWKDFHFERIQFDPAEMSV
This sequence was then submitted to the HHpred server using default settings. The result appeared as shown below.
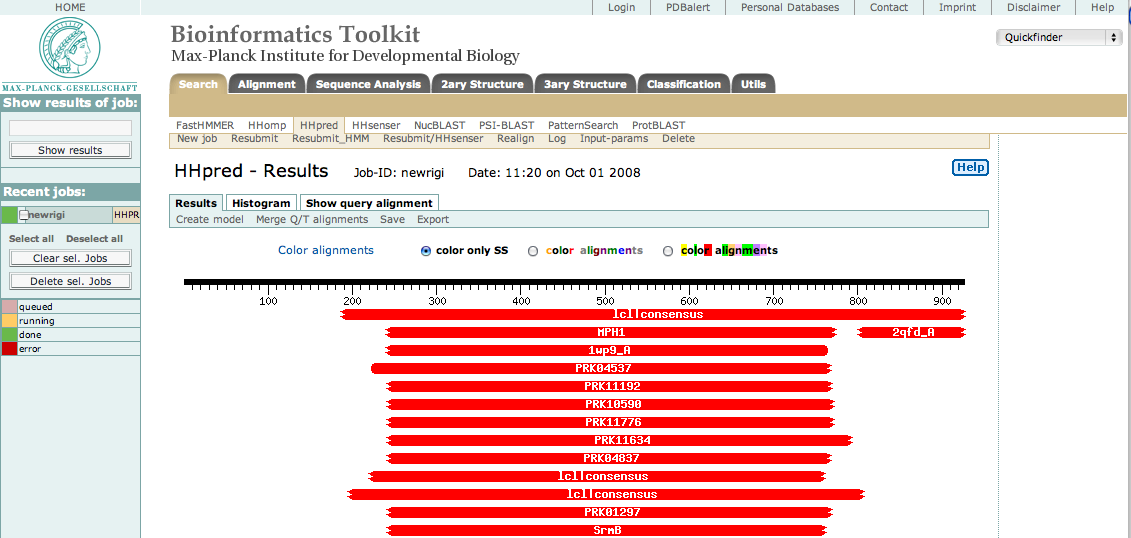
As you can see, there are many hits to the central region approximately from res 250 to 775. This is the helicase domain. By selecting only these residues in our original sequence, resubmitting, and then selecting the best hits, we were able to identify three sub-domains consisting of the following segments
>ATPase-like 1 ECQNLSQNPGPPSEASSNNLHSPLKPRNYQLELALPAKKGKNTIICAPTGCGKTFVSLLICEHRLKKFPCGQKGKVVFFANQIPVYEQQAT VFSRYFERLGYNIASISGATSDSVSVQHIIEDNDIIILTPQILVNNLNNGAIPSLSVFTLMIFDECHNTSKNHPYNQIMFRYLDHKLGESRDPL PQVVGLTAS >Helical VGVGDAKTAEEAMQHICKLCAALDASVIATVRDNVAELEQVVYKPQKISRKVASRTSNTFKCIISQLMKETEKLAKDVSEELGKLFQIQN REFGTQKYEQWIVGVHKACSVFQMADKEEESRVCKALFLYTSHLRKYNDALIISEDAQMTDALNYLKAFFHDVREAAFDETERELTRRF EEKLEELEKVS > ATPase-like 2 RGPSNENPKLRDLYLVLQEEYHLKPETKTILFVKTRALVDALKKWIEENPALSFLKPGILTGRGRTNRATGMTLPAQKCVLEAFRASGDNNI LIATSVADEGIDIAECNLVILYEYVGNVIKMIQTRGRGRARDSKCFLL
Note that the following instructions refer to September 2008 HHpred server results. These results are subject to change.
We then built a model for the second 'ATPase-like' domain by selecting the 'Create model' button at the top of the page, and selecting the PDB entry 2db3 as the template. Note that this template is not the default choice. It was selected because the sequence identity (25%) was higher than that of 1wp9 (19%). You will have to follow the instructions to obtain a MODELLER license key. This resulting model can be found here.
Note that there is one complication: MODELLER does not, by default, add a chain ID to the PDB file. SeSAW, on the other hand, requires a chain ID. To get around this problem, we have prepared a simple web-based utility that adds a chain ID to a PDB-formatted file. The utility can be found here. Add a chain ID to the PDB-formatted file by using this utility.
2. We then can submit the modeled structure to SeSAW as described above. Remember to input the template PDB ID (2db3 in this case). We will assume that the submission process was successful, and skip to analysis of the results.
3. The results table for the 2db3 model is shown below. The top hits are all helicases, which gives us some confidence in our model.
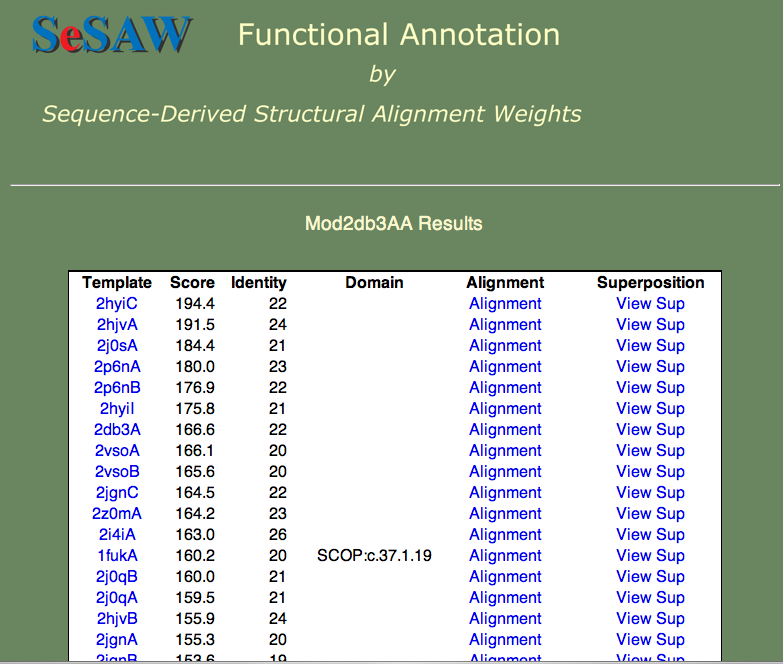
Helicases stabilize the unwinding of double-stranded nucleaotides (DNA or RNA). Given the fact that nucleotides are negatively charged, we expect to see positively charged amino acids (R (Arginine), K (Lysine), or H (Histidine)) at the nucleotide binding site. Can you find candidate residues by looking at the high-scoring residues in a few of the high-scoring alignments? In the figure below, the top-scoring alignment to the 'exon junction complex with a trapped DEAD-box helicase' (PDB ID 2hyi) is shown.

If you look at the last 20 residues in the model you will see that it contains 4 Arginine residues and one Lysine residue. Of these, R 116 appears in a number of the alignments. What other two residues also appear that are close to R 116. Hint:: close does not necessarily mean close in residue number. To find the 'nearby' residues you will have to view the superposition.
Note also that the particular region we are looking at contains at least one gap in the alignment and the secondary structure is not completely regular (i.e., the distinction between loop and helix is not obvious). In other words, this region of interest is not trivial to model. Therefore, we must be careful about our functional interpretation of the data. These types of modeling problems occur frequently at binding sites. Can you think of a reason why? A binding site must often be flexible in order to perform its function. For example, it is common for a protein to assume different conformations in the bound and unbound state. Our templates represent an ensemble of different conformations. They also bind different nucleotides than our query. At this point we can say that the C-terminal region is probably involved in RNA binding, but more experimental evidence (i.e., mutational analysis, along with direct structural evidence) is necessary to pin down the exact role of this region in RNA recognition.
Toll-like receptor 9 TIR domain
The sequence of the Toll/interleukin-1 receptor from human Toll-like receptor 9 (TLR-9 TIR domain) is shown below.
DAFVVFDKTQSAVADWVYNELRGQLEECRGRWALRLCLEERDWLPGKTLFENLWASVYGSRKTLFVLAHTDRVSGLLRASFLLAQQRLLE DRKDVVVLVILSPDGRRSRYVRLRQRLCRQSVLLWPHQPSGQRSFWAQLGMALTRDNHHFYNRNFCQGPTAE
TLRs are involved in the innate immune response and contain an receptor domain, followed by a transmembrane helix, and, at the C-terminus, a signaling domain. The signaling domain binds to other signaling proteins inside the cell thereby warning the cell of possible infection. Here we are interested in finding out what parts on the TIR domain from TLR-9 are likely to be involved in such protein-protein interactions.
A model was built on human TLR-1 (1fyv), with a sequence identity of 27%, following the default HHpred procedure as above. The highest-scoring residue pairs in the SeSAW alignment included a number of conserved hydrophobic residues (W16, W125, Y110, F135, W136), along with prolines (P45,P103,P126) that are characteristic of TIR domains.
<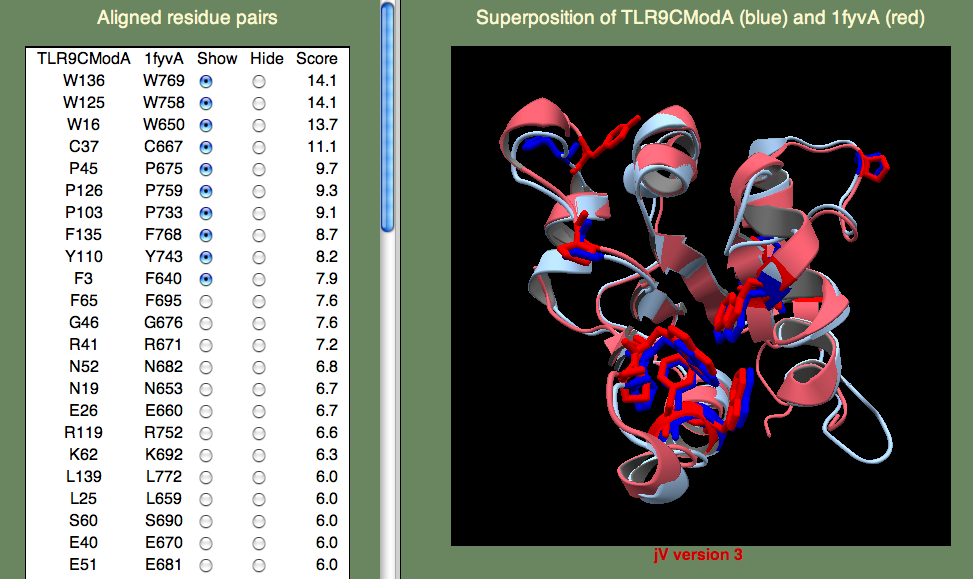
If you have time to take a look at the primary reference for 1fyv you will find that the TIR domains from TLR-9 and TLR-1 share five residues in a conserved patch that includes the ‘cBB loop,’ thought to be involved in receptor signaling. These identical residues (E40, R41, P45, G46, K47) score highly. Note that we don't expect the TIR binding sites of TLR-9 and TLR-1 to be identical. Their differences are consistent with a different signal cascades initiated by these two innate immune response receptors: TLR-9 detects unmethylated CpG DNA and TLR-1 detects lipoproteins derived from bacteria.
Alternative oxidase from Blastocystis
Alternative oxidase (AOX) is an iron binding protein found in the mitochondria. Blastocystis is an intestinal parasite that contains unusual mitochondria-like organelles that don't require oxygen (anaerobic), although their function is not well known.
MFPILSRVFFKREAVVFRGFSVSSYEQFIDKECISKALNKKPNEHYHIFSTRYHSSNREYLTILESCWGEQPKRHPKGVSDRVASGIVNALFKIGN AYFRENYILRAVFLESVASIPGLVCSNLHHLRCLRRLQPDSWIKPLVDEAENERMHLLAVRTYTKLTAVQKLFIRITQFSFVTLFSFLFVFAPRTS HRLVGFLEEHAVDSYTEMIRRIDSNTLENRPATQITKDYWGLPEDATLRDALLVIRADEADHRLVNHSLGDAYDKKTPVSVKKWYAGCAFP VNLHEPFGPYMDFSKYGATKA
The sequence identity to known folds is very low, so AOX is a tricky case. Here we want to try several models and pick out the best one based on the alignment of the known functionally important residues. There is a pair of iron-binding motifs that you want to be are are present in you final model. The motif is E...ExxH which means, there is a Glutamic acid, followed by a variable length of residues, followed by another Glutamic acid, followed by two residues, followed by a Histadine. This sounds rather arbitrary, but remember that residues bind iron, so you expect to find them close together. Also remember that the motif occurs twice, so you are looking for two such patterns, and these patterns should be close to one another as well. To save time, we have provided a model for you here.
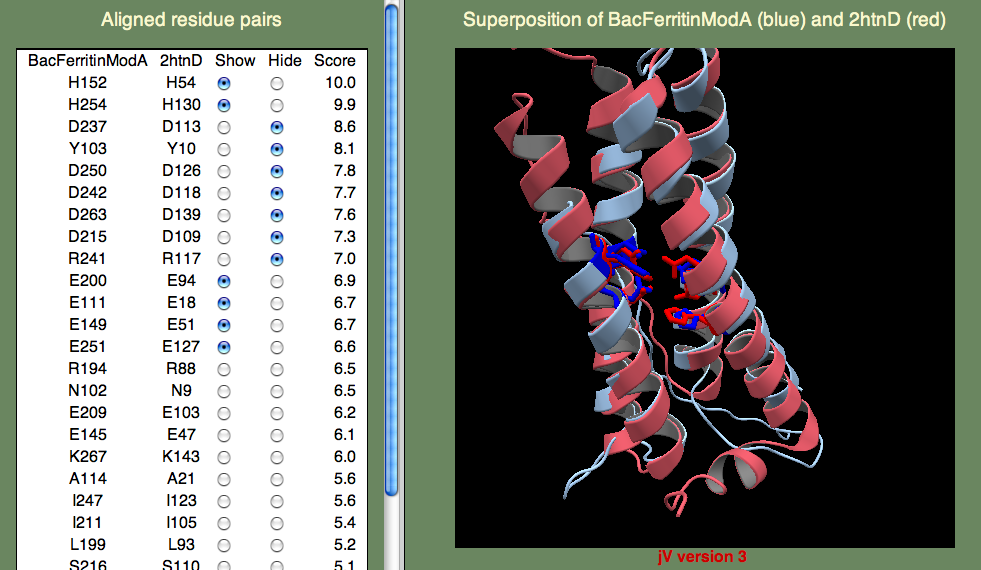
Below we show the residues in question in the superposition on template 2htn. Remember that 2htn is a different protein (bacterioferritin) and so we don't expect that AOX and 2htn are structurally identical. In particular, AOX has a membrane-bound helix that is not present in bacterioferritin. Can you guess where this helix might be? Hint, the helix is not a transmembrane helix, but rather a helix that enters and leaves the membrane parallel to the surface, without crossing it. To find this region you will have to consult the secondary structure prediction of the query (shown below).




