 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4qag: Structure of a dihydroxycoumarin active-site inhibitor in complex... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4qag | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of a dihydroxycoumarin active-site inhibitor in complex with the RNASE H domain of HIV-1 reverse transcriptase | |||||||||
 Components Components | Reverse transcriptase/ribonuclease H | |||||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / RNASE H INHIBITOR / STRUCTURE-BASED DRUG DESIGN / ACTIVE SITE / TRANSFERASE / DIHYDROXYCOUMARIN ANALOGS / DIHYDROXY-BENZOPYRONE DERIVATIVES / DIVALENT CATION CHELATOR / AIDS / REVERSE TRANSCRIPTASE / PROTEIN-INHIBITOR COMPLEX / HYDROLASE-HYDROLASE INHIBITOR complex | |||||||||
| Function / homology |  Function and homology information Function and homology informationHIV-1 retropepsin / symbiont-mediated activation of host apoptosis / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / host multivesicular body / DNA integration / viral genome integration into host DNA / RNA-directed DNA polymerase / establishment of integrated proviral latency ...HIV-1 retropepsin / symbiont-mediated activation of host apoptosis / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / host multivesicular body / DNA integration / viral genome integration into host DNA / RNA-directed DNA polymerase / establishment of integrated proviral latency / viral penetration into host nucleus / RNA stem-loop binding / symbiont-mediated suppression of host gene expression / RNA-directed DNA polymerase activity / host cell / RNA-DNA hybrid ribonuclease activity / Transferases; Transferring phosphorus-containing groups; Nucleotidyltransferases / viral nucleocapsid / aspartic-type endopeptidase activity / DNA recombination / DNA-directed DNA polymerase / Hydrolases; Acting on ester bonds / DNA-directed DNA polymerase activity / symbiont entry into host cell / lipid binding / host cell nucleus / host cell plasma membrane / structural molecule activity / virion membrane / proteolysis / DNA binding / zinc ion binding / membrane Similarity search - Function | |||||||||
| Biological species |   Human immunodeficiency virus type 1 Human immunodeficiency virus type 1 | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.712 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.712 Å | |||||||||
 Authors Authors | Himmel, D.M. / Ho, W.C. / Arnold, E. | |||||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2014 Title: Structure of a Dihydroxycoumarin Active-Site Inhibitor in Complex with the RNase H Domain of HIV-1 Reverse Transcriptase and Structure-Activity Analysis of Inhibitor Analogs. Authors: Himmel, D.M. / Myshakina, N.S. / Ilina, T. / Van Ry, A. / Ho, W.C. / Parniak, M.A. / Arnold, E. #1: Journal: Structure / Year: 2009Title: Structure of HIV-1 Reverse Transcriptase with the Inhibitor beta-Thujaplicinol Bound at the RNase H Active Site Authors: Himmel, D.M. / Maegley, K.A. / Pauly, T.A. / Bauman, J.D. / Das, K. / Dharia, C. / Clark Jr., A.D. / Ryan, K. / Hickey, M.J. / Love, R.A. / Hughes, S.H. / Bergqvist, S. / Arnold, E. #2: Journal: J.Med.Chem. / Year: 2011Title: Synthesis, Activity, and Structural Analysis of Novel ALPHA-HYDROXYTROPOLONE INHIBITORS OF HUMAN IMMUNODEFICIENCY VIRUS REVERSE TRANSCRIPTASE-ASSOCIATED RIBONUCLEASE H Authors: Chung, S. / Himmel, D.M. / Jiang, J. / Wojtak, K. / Bauman, J.D. / Rausch, J.W. / Wilson, J.A. / Beutler, J.A. / Thomas, C.J. / Arnold, E. / Le Grice, S.F.J. #3: Journal: Proc.Natl.Acad.Sci.USA / Year: 2008Title: HIGH-RESOLUTION STRUCTURES OF HIV-1 REVERSE TRANSCRIPTASE/TMC278 COMPLEXES: STRATEGIC FLEXIBILITY EXPLAINS POTENCY AGAINST RESISTANCE MUTATIONS Authors: Das, K. / Bauman, J.D. / Clark Jr., A.D. / Frenkel, Y.V. / Lewi, P.J. / Shatkin, A.J. / Hughes, S.H. / Arnold, E. #4: Journal: ACS CHEM.BIOL. / Year: 2006Title: HIV-1 REVERSE TRANSCRIPTASE STRUCTURE WITH RNASE H INHIBITOR DIHYDROXY BENZOYL NAPHTHYL HYDRAZONE BOUND AT A NOVEL SITE Authors: Himmel, D.M. / Sarafianos, S.G. / Dharmasena, S. / Hossain, M.M. / McCoy-Simandle, K. / Ilina, T. / Clark Jr., A.D. / Knight, J.L. / Julias, J.G. / Clark, P.K. / Krogh-Jespersen, K. / Levy, ...Authors: Himmel, D.M. / Sarafianos, S.G. / Dharmasena, S. / Hossain, M.M. / McCoy-Simandle, K. / Ilina, T. / Clark Jr., A.D. / Knight, J.L. / Julias, J.G. / Clark, P.K. / Krogh-Jespersen, K. / Levy, R.M. / Hughes, S.H. / Parniak, M.A. / Arnold, E. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4qag.cif.gz | 131.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4qag.ent.gz | 100.6 KB | Display | PDB format |
| PDBx/mmJSON format | 4qag.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 4qag_validation.pdf.gz | 453 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 4qag_full_validation.pdf.gz | 456.4 KB | Display | |
| Data in XML | 4qag_validation.xml.gz | 14.7 KB | Display | |
| Data in CIF | 4qag_validation.cif.gz | 20.1 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qa/4qagftp://data.pdbj.org/pub/pdb/validation_reports/qa/4qag | HTTPS FTP |
-Related structure data
| Related structure data |  3ig1S S: Starting model for refinement |
|---|---|
| Similar structure data |








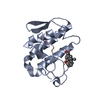





-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 14714.667 Da / Num. of mol.: 2 / Fragment: UNP residues 1024-1156 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Human immunodeficiency virus type 1 / Gene: gag-pol / Plasmid: pLysS / Production host:  References: UniProt: P03366, RNA-directed DNA polymerase, DNA-directed DNA polymerase, retroviral ribonuclease H, exoribonuclease H #2: Chemical | ChemComp-MN /   Mass: 54.938 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mn#3: Chemical | 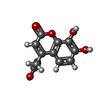  Mass: 236.178 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C11H8O6 Mass: 236.178 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C11H8O6#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 176 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 176 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.88 Å3/Da / Density % sol: 57.27 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 8.2 Details: 100 mM Bicine pH 8.2, 10 mM Manganese Sulfate, 1 mM Sodium Azide, 9% PEG 3350, combined with equal volume of 10 mM Tris pH 8.0, 75 mM NaCl, 20 mg/mL (1.34 mM) RNase H, VAPOR DIFFUSION, ...Details: 100 mM Bicine pH 8.2, 10 mM Manganese Sulfate, 1 mM Sodium Azide, 9% PEG 3350, combined with equal volume of 10 mM Tris pH 8.0, 75 mM NaCl, 20 mg/mL (1.34 mM) RNase H, VAPOR DIFFUSION, HANGING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X25 / Wavelength: 1.1 / Wavelength: 1.1 Å / Beamline: X25 / Wavelength: 1.1 / Wavelength: 1.1 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Jun 3, 2010 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.1 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Redundancy: 7.8 % / Number: 272419 / Rmerge(I) obs: 0.088 / Χ2: 1 / D res high: 1.71 Å / D res low: 40 Å / Num. obs: 34856 / % possible obs: 98.5 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Diffraction reflection shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.71→40 Å / Num. all: 35387 / Num. obs: 34856 / % possible obs: 98.5 % / Observed criterion σ(I): -0.4 / Redundancy: 7.8 % / Biso Wilson estimate: 34.341 Å2 / Rmerge(I) obs: 0.088 / Rsym value: 0.088 / Χ2: 1.003 / Net I/σ(I): 27.1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
-Phasing
| Phasing | Method: molecular replacement | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MR | Rfactor: 38.45 / Model details: Phaser MODE: MR_AUTO
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 3IG1 Resolution: 1.712→28.565 Å / SU ML: 0.23 / Isotropic thermal model: ANISOTROPIC / Cross valid method: THROUGHOUT / σ(F): 0 / Phase error: 26.88 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 159.53 Å2 / Biso mean: 56.6933 Å2 / Biso min: 30.35 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error free: 0.23 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.712→28.565 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 7
|
