 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1wyk | ||||||
|---|---|---|---|---|---|---|---|
| Title | SINDBIS VIRUS CAPSID PROTEIN (114-264) | ||||||
 Components Components | SINDBIS VIRUS CAPSID PROTEIN | ||||||
 Keywords Keywords | Viral protein / hydrolase / COAT PROTEIN / SINDBIS / VIRUS / PROTEINASE / ALPHAVIRUS / CAPSID / DIOXANE | ||||||
| Function / homology |  Function and homology information Function and homology informationtogavirin / T=4 icosahedral viral capsid / host cell endoplasmic reticulum / channel activity / monoatomic ion transmembrane transport / symbiont-mediated suppression of host toll-like receptor signaling pathway / host cell Golgi apparatus / entry receptor-mediated virion attachment to host cell / serine-type endopeptidase activity / fusion of virus membrane with host endosome membrane ...togavirin / T=4 icosahedral viral capsid / host cell endoplasmic reticulum / channel activity / monoatomic ion transmembrane transport / symbiont-mediated suppression of host toll-like receptor signaling pathway / host cell Golgi apparatus / entry receptor-mediated virion attachment to host cell / serine-type endopeptidase activity / fusion of virus membrane with host endosome membrane / symbiont entry into host cell / host cell nucleus / host cell plasma membrane / virion membrane / structural molecule activity / proteolysis / RNA binding Similarity search - Function | ||||||
| Biological species |  Sindbis virus Sindbis virus | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Lee, S. / Kuhn, R.J. / Rossmann, M.G. | ||||||
 Citation Citation | Journal: Proteins / Year: 1998 Title: Probing the potential glycoprotein binding site of sindbis virus capsid protein with dioxane and model building. Authors: Lee, S. / Kuhn, R.J. / Rossmann, M.G. #1: Journal: Structure / Year: 1996Title: Identification of a Protein Binding Site on the Surface of the Alphavirus Nucleocapsid and its Implication in Virus Assembly Authors: Lee, S. / Owen, K.E. / Choi, H.K. / Lee, H. / Lu, G. / Wengler, G. / Brown, D.T. / Rossmann, M.G. / Kuhn, R.J. #2: Journal: Nature / Year: 1991Title: Structure of Sindbis Virus Core Protein Reveals a Chymotrypsin-Like Serine Proteinase and the Organization of the Virion Authors: Choi, H.K. / Tong, L. / Minor, W. / Dumas, P. / Boege, U. / Rossmann, M.G. / Wengler, G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1wyk.cif.gz | 176.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1wyk.ent.gz | 140.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1wyk.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/wy/1wykftp://data.pdbj.org/pub/pdb/validation_reports/wy/1wyk | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj
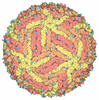

- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 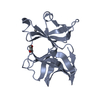
| ||||||||||||||||||||
| 2 | 
| ||||||||||||||||||||
| 3 | 
| ||||||||||||||||||||
| 4 | 
| ||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
|
-Components
| #1: Protein | Mass: 16676.828 Da / Num. of mol.: 4 / Fragment: C-TERMINAL DOMAIN Source method: isolated from a genetically manipulated source Source: (gene. exp.) Sindbis virus / Genus: Alphavirus / Strain: SA-AR 86 / Cell line: BL21 / Gene: POTENTIAL / Plasmid: BL21 / Species (production host): Escherichia coli / Production host:  References: UniProt: P27285, Hydrolases; Acting on peptide bonds (peptidases); Serine endopeptidases #2: Chemical | ChemComp-FOR /   Mass: 30.026 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: CH2O Mass: 30.026 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: CH2O#3: Chemical | ChemComp-DIO / 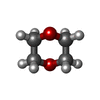  Mass: 88.105 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C4H8O2 Mass: 88.105 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C4H8O2#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 469 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 469 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2 Å3/Da / Density % sol: 38.5 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6.5 Details: 20-26%(W/V) PEG 8000, 100 MM SODIUM CACODYLATE, PH6.5, 150MM SODIUM ACETATE, 6%(V/V) DIOXANE | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 7.6 / Method: vapor diffusion, hanging drop / Details: used to seeding | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 110 K |
|---|---|
| Diffraction source | Wavelength: 1.5418 |
| Detector | Type: RIGAKU / Detector: IMAGE PLATE / Date: Oct 26, 1995 / Details: BENT FOCUSING MIRROR |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2→30 Å / Num. obs: 28851 / % possible obs: 78.3 % / Observed criterion σ(I): 1 / Redundancy: 1.9 % / Biso Wilson estimate: 15.2 Å2 / Rsym value: 0.028 / Net I/σ(I): 16 |
| Reflection shell | Resolution: 2→2.05 Å / Redundancy: 1.6 % / Mean I/σ(I) obs: 6 / Rsym value: 0.089 / % possible all: 28.8 |
| Reflection | *PLUS Num. measured all: 53873 / Rmerge(I) obs: 0.028 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: SINDBIS VIRUS CAPSID PROTEIN MUTANT (S215A, 106-266) IN TRICLINIC CRYSTAL Resolution: 2→8 Å / Rfactor Rfree error: 0.0072 / Data cutoff high absF: 304 / Data cutoff low absF: 9.7 / σ(F): 1
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 15.6 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati d res low obs: 30 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | NCS model details: MOL1 AND MOL4, MOL2 AND MOL3 WERE RESTRAINED IN A PAIRWISE MANNER DURING REFINEMENT. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.03 Å / Rfactor Rfree error: 0.074 / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
