 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
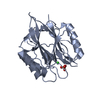
Yorodumi- PDB-4lq6: Crystal structure of Rv3717 reveals a novel amidase from M. tuber... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4lq6 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Rv3717 reveals a novel amidase from M. tuberculosis | ||||||
 Components Components | N-acetymuramyl-L-alanine amidase-related protein | ||||||
 Keywords Keywords |  HYDROLASE / Amidase / cell wall hydrolase HYDROLASE / Amidase / cell wall hydrolase | ||||||
| Function / homology |  Function and homology informationN-acetylmuramoyl-L-alanine amidase activity / peptidoglycan catabolic process / metal ion binding Function and homology informationN-acetylmuramoyl-L-alanine amidase activity / peptidoglycan catabolic process / metal ion bindingSimilarity search - Function | ||||||
| Biological species |   Mycobacterium tuberculosis (bacteria) Mycobacterium tuberculosis (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 1.68 Å | ||||||
 Authors Authors | Kumar, A. / Kumar, S. / Kumar, D. / Mishra, A. / Dewangan, R.P. / Shrivastava, P. / Ramachandran, S. / Taneja, B. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2013 Title: The structure of Rv3717 reveals a novel amidase from Mycobacterium tuberculosis. Authors: Kumar, A. / Kumar, S. / Kumar, D. / Mishra, A. / Dewangan, R.P. / Shrivastava, P. / Ramachandran, S. / Taneja, B. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4lq6.cif.gz | 98.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4lq6.ent.gz | 81 KB | Display | PDB format |
| PDBx/mmJSON format | 4lq6.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/lq/4lq6ftp://data.pdbj.org/pub/pdb/validation_reports/lq/4lq6 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4hjn |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 22696.316 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Mycobacterium tuberculosis (bacteria) / Gene: MT3820, Rv3717 / Production host: Escherichia coli (E. coli) / References: UniProt: O69684 |
|---|
-Non-polymers , 5 types, 200 molecules 




| #2: Chemical | ChemComp-ZN /  Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn | ||||
|---|---|---|---|---|---|
| #3: Chemical | ChemComp-SO4 / Sulfate Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 | ||||
| #4: Chemical | ChemComp-CL / Chloride Mass: 35.453 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Cl#5: Chemical |  Mass: 195.078 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Pt Mass: 195.078 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Pt#6: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 191 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.77 Å3/Da / Density % sol: 55.67 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 7.5 Details: Ammonium sulfate, pH 7.5, VAPOR DIFFUSION, SITTING DROP, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: BM14 / Wavelength: 1.0714 Å / Beamline: BM14 / Wavelength: 1.0714 Å |
| Detector | Type: MARMOSAIC 225 mm CCD / Detector: CCD / Date: May 16, 2012 |
| Radiation | Monochromator: silicon mirrors / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.0714 Å / Relative weight: 1 |
| Reflection | Resolution: 1.68→29.52 Å / Num. all: 29381 / Num. obs: 29381 / % possible obs: 100 % / Observed criterion σ(F): 3 / Observed criterion σ(I): 0 / Biso Wilson estimate: 21.46 Å2 |
| Reflection shell | Resolution: 1.68→1.74 Å / % possible all: 3.23 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAD / Resolution: 1.68→21.09 Å / Cor.coef. Fo:Fc: 0.9601 / Cor.coef. Fo:Fc free: 0.9501 / SU R Cruickshank DPI: 0.079 / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 23.58 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.177 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.68→21.09 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.68→1.74 Å / Total num. of bins used: 15
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 10.6726 Å / Origin y: 40.9984 Å / Origin z: 45.2963 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection details: { A|2 - 214 } |
